


血友病:一种X连锁隐性遗传病
血友病是一种典型的X染色体连锁的隐性遗传性出血性疾病,其核心病因在于特定凝血因子的缺乏或功能异常。根据所缺乏的凝血因子种类,血友病主要分为两大类:血友病A,由凝血因子VIII(FVIII)缺乏引起;以及血友病B,由凝血因子IX(FIX)缺乏所致。临床上,患者常常表现为自发性或轻微创伤后难以控制的出血,尤其是在关节、肌肉和深部组织。长期的反复出血不仅会导致严重的关节畸形和功能障碍,还可能引发危及生命的大出血,对患者的生活质量和预期寿命构成严重威胁。
全球与中国的血友病流行病学特征
根据世界血友病联盟(WFH)2022年发布的全球调查报告,血友病的患病率在全球范围内存在显著的地区差异。尽管中国的患病率(每10万人中约2-3例)低于许多发达国家,但鉴于其庞大的人口基数,中国已成为全球拥有最多血友病患者的国家,登记在册的患者人数超过32,000人。然而,这一数字很可能低估了实际情况,因为许多轻症患者或居住在医疗资源匮乏地区的患者可能未能得到及时诊断和统计。因此,提高公众和医务人员对血友病的认知,加强诊断和报告体系,对于全面了解中国的血友病负担至关重要。
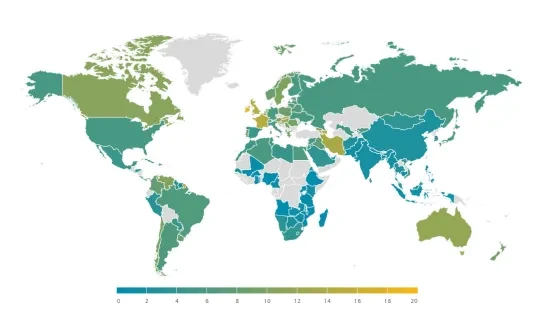
图1. 2022年血友病每10万人患病率
血友病的分子遗传学基础:F8与F9基因的关键作用
现代分子遗传学研究表明,约86%的血友病病例是由特定的基因突变所致。这些突变主要集中在两个位于X染色体上的基因:

图2. 近年来血友病发生与基因突变相关性
- F8基因与血友病A:F8基因位于X染色体长臂的末端区域(Xq28),负责编码凝血因子VIII。该基因的突变形式多样,包括基因倒位、大片段缺失、点突变等。罕见病数据中心(RDDC)的资料显示,目前已发现超过500种与血友病A相关的F8基因致病性突变。这些突变直接影响FVIII的合成和功能,是导致血友病A的根本原因。
- F9基因与血友病B:F9基因同样位于X染色体上(Xq26.3-27.2),编码凝血因子IX。与F8基因类似,F9基因的突变也会导致FIX蛋白的合成障碍或功能缺陷,从而引发血友病B。RDDC数据库已收录超过300种与血友病B相关的F9基因突变。对这些突变的深入研究,为血友病的精确诊断、携带者筛查和遗传咨询提供了科学依据。
动物模型在血友病研究中的核心价值
由于人类和小鼠在基因组上具有高度的同源性,小鼠模型已成为血友病研究中不可或缺的工具。通过先进的TurboKnockout-Pro技术,可以高效、精确地构建F8或F9基因敲除的小鼠模型。这些模型能够很好地模拟人类血友病的出血表型,例如关节自发性出血和创伤后出血时间延长等。利用这些动物模型,科研人员可以深入探究血友病的发病机制,并对新型治疗方法(如基因治疗、抗体药物等)的有效性和安全性进行临床前评估,极大地加速了研究成果向临床应用的转化。
基因治疗:引领血友病治疗的革命性突破
在过去的几十年里,血友病的标准治疗方法是凝血因子替代疗法,即定期静脉注射浓缩的凝血因子。尽管这种方法有效,但存在诸多不便,如需要频繁注射、可能产生抑制物以及治疗成本高昂等。基因治疗的出现,为从根本上治愈血友病带来了希望。
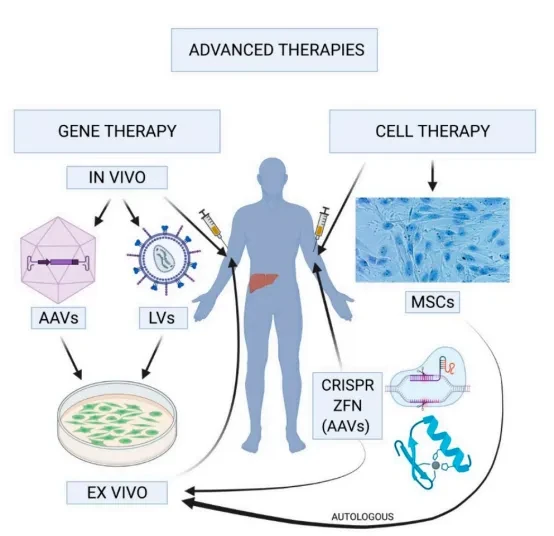
图3. 血友病基因治疗工作原理
基因治疗的核心思想是通过特定的病毒载体(如腺相关病毒AAV或慢病毒LV),将正常的F8或F9基因导入患者体内的特定细胞(主要是肝细胞),使其能够长期、稳定地表达功能性凝血因子。这种“一次性治疗,终身受益”的模式,有望彻底改变血友病的治疗格局。目前,全球范围内已有多种针对血友病B的AAV基因治疗药物(如Etranacogene dezaparvovec)获批上市,其临床试验结果显示,单次注射后患者体内的FIX水平能够恢复到接近正常的水平,显著减少甚至完全摆脱了出血事件。针对血友病A的基因治疗研究也取得了重大进展,多种药物正处于后期临床试验阶段,预示着血友病治疗即将进入一个全新的、以基因治疗为主导的时代。
内容来源与免责声明
本文基于公开的科学文献和数据库信息编写,旨在提供专业、准确的行业洞察。内容仅供科研参考,不作为临床诊断或治疗的依据。




