

腓骨肌萎缩症(CMT)——纤细的“鹤腿”
疾病概述
很多人都想拥有一双又细又长的腿,他们节食、跑步,不断地尝试各种方法,却一次又一次地无功而返。然而,大概在每10000人中就有4个人,他们不用减肥,小腿却会自己悄悄“瘦”下来,像鹤腿一样,他们就是“腓骨肌萎缩症”患者。

图1.图源来自网络
腓骨肌萎缩症(Charcot-Marie-Tooth,CMT)病是一组由不同致病基因引起的临床表型基本相似的遗传性周围神经病,不同人群中CMT的发病率略有不同,约在17/10000~40/100000之间。自法国医师Charcot、Marie和英国医师Tooth于1886年首先报道腓骨肌萎缩症后,以他们姓氏Charcot-Marie-Tooth命名腓骨肌萎缩症,并将之归为周围神经病。大多数类型腓骨肌萎缩症会同时累及运动和感觉神经元,因此Dyck和Lambert于1968年提出新的名称遗传性运动感觉神经病(hereditary motor and sensory neuropathy,HMSN)命名腓骨肌萎缩症[1]。
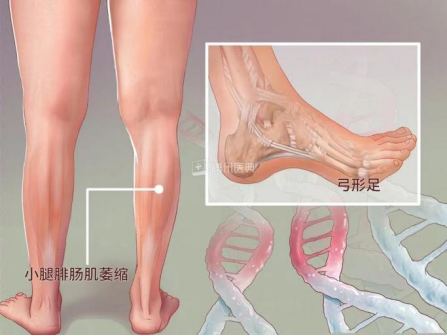
图2.图源来自腾讯医典
基因分型
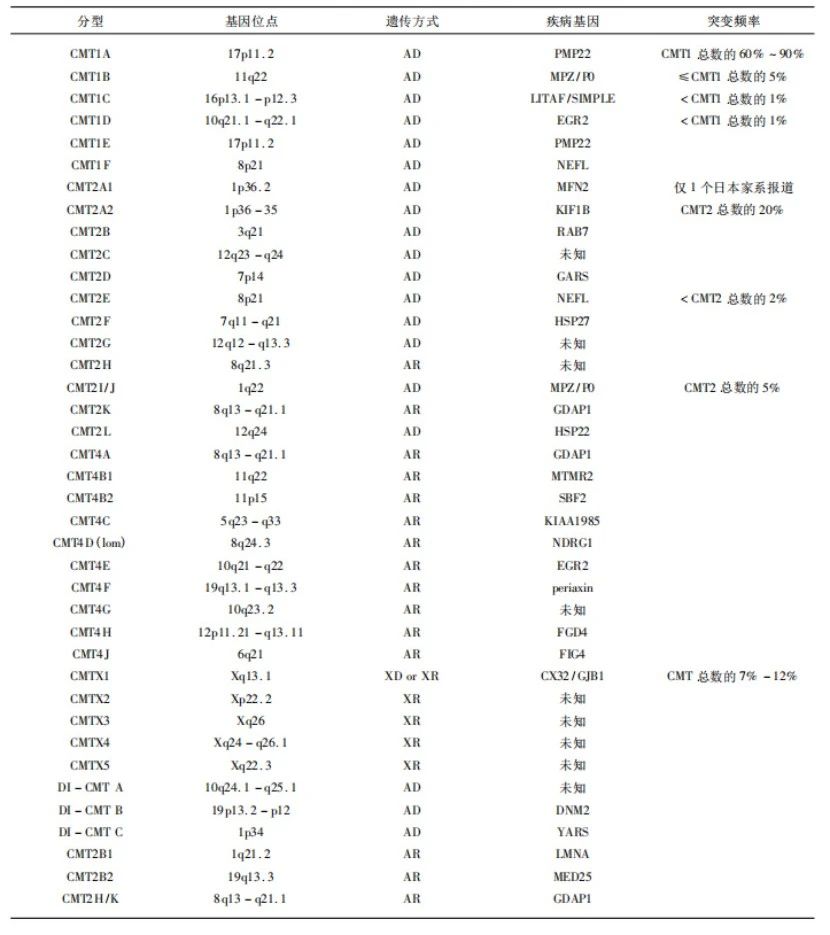
CMT的遗传方式以常染色体显性遗传最多见,见于大部分CMT1和CMT2家系患者,其次为X连锁显性遗传;常染色体隐性遗传较少见,而散发病例并不少见。根据神经电生理、病理特点,CMT可分为脱髓鞘型(CMT1型)、轴索型(CMT2型)和中间型(ICMT型)[2]。近年来发现一些脱髓鞘与轴突变性共存的中间型常染色体显性遗传CMT(DI-CMT),依据CMT的遗传位点和疾病基因可将CMT分为CMT1、CMT2、CMT4、CMTX和DI-CMT5类(见表1)[3]。
表1.CMT基因型及疾病基因一览表[3]
发病机制
外周祌经系统由神经元细胞、施旺细胞和成纤维细胞组成。施旺细胞和神经元细胞能够相互交换分子信号来调节周围神经的生存和分化,在CMT患者中,这些细胞间的信号可能被扰乱。
髓鞘是包裹在有鞘神经纤维轴突上的管状外膜,由施旺细胞质膜卷折形成,主要起到支持轴突、绝缘和帮助神经细胞更快地进行信号传导的作用,当出现显著的施旺细胞病变导致髓鞘脱失时,神经传导速度减慢。施旺细胞是由基膜环绕的,而基膜中的细胞外基质(ECM)分子和他们的受体对施旺细胞的功能起到了重要的调节作用,ECM分子不但提供支持作用,还能触发细胞内的信号转导过程,例如调节施旺细胞包裹轴突这一过程的发生。ECM信号通路主要包括,胶原蛋白、层黏连蛋白、整合素、糖蛋白等,这些蛋白可以激活下游的受体包括FAK,小RhoGTP酶,ILK以及PI3K/Aktp信号通路。于此同时,由于施旺细胞起到了支持和帮助轴突生长的作用,施旺细胞脱髓鞘也可以引起轴突结构和生长的异常,这种情况下会同时导致髓鞘和轴突受损。
轴突变性主要由三种机制导致:轴突运输功能损伤,线粒体功能障碍以及钙离子的轴浆内流增加。轴突变性是一种长度依赖性病变,神经纤维越长,维持长距离轴浆运输所需线粒体及其所产生的能量越多,因此CMT2首先影响四肢远端神经纤维,并逐步导致其所支配的肌肉发生萎缩。现有的研究通常认为CMT的发生与轴突运输、线粒体功能、囊泡周期循环、细胞骨架的稳定性、离子通道释放功能的异常相关[4]。
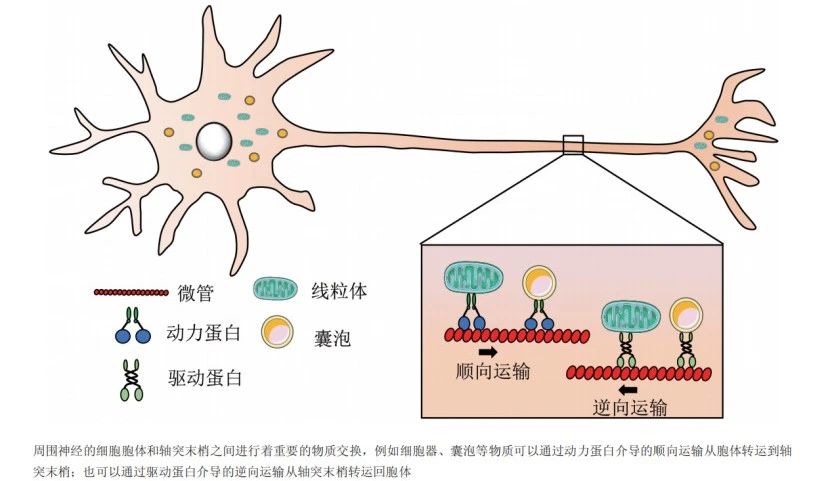
图3.轴突运输[4]
临床表现
CMT患者多在10岁前发病,少数患者在成年发病,病程进展缓慢。CMT的临床表现具有一些相似之处,常表现为进行性对称性肢体远端肌无力和肌萎缩,部分患者可伴远端感觉减退或缺失、骨骼畸形、腱反射减弱或缺失。肌萎缩常由下肢开始逐渐发展到上肢,大腿下1/3以下肌肉无力和萎缩,形成“鹤腿”或倒置酒瓶样畸形,行走和跑步困难,跨阈步态。
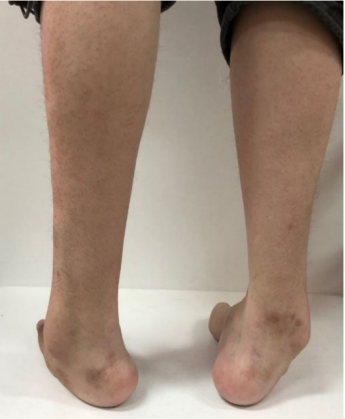
图4.腓骨肌萎缩症患者骨骼畸[6]
手部骨间肌和大小鱼际肌无力和萎缩,出现爪形手或猿手畸形,肌萎缩一般不超过肘关节以上,手不能做出精细的动作;四肢近端肌肉萎缩较为少见,仅出现在一些症状较重的患者中;下肢比上肢更易出现末梢型感觉障碍,通常痛温觉和振动觉均减退,位置觉较少受损,也是先累及足部,再向小腿延伸,然后累及手部,腱反射减弱或缺失,可伴自主神经功能障碍和营养障碍。CMT常伴弓形足、脊柱侧弯等骨骼畸形,少数患者可先出现扁平足,然后转变为弓形足。晚期发病的CMT患者往往无足部骨骼畸形。其他常见的症状和体征包括肌肉痛性痉挛、双足发冷、发绀和过度角质化等,发病极早的病例可导致肌张力低下,运动发育迟缓,踮脚走路。
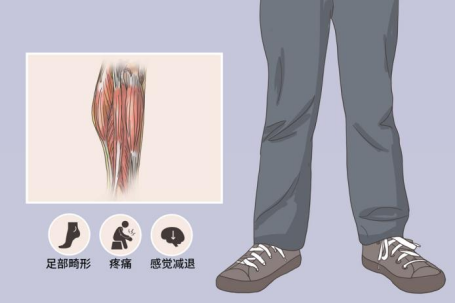
图5.腓骨肌萎缩症患者常见临床表现[7]
除上述CMT共同的临床特征外,一些类型CMT可出现特有的神经受累症状。如CMT1D可出现脑神经受损,CMT1X可出现中枢神经受损,CMT2A可出现视神经萎缩,CMT2C可出现声带麻痹及呼吸受累,CMT2D上肢受累严重,CMT2J可出现Adie’s瞳孔,CMT4B2可出现早发性青光眼,CMT4C可出现重度脊柱侧弯,CMT4F感觉缺失明显,HMNSR可出现听力丧失,DI-CMTB可出现神经痛。但不是所有该型CMT患者均有这些特征症状。在同一CMT家系中,不同个体出现的临床症状轻重有所不一,有的个体出现典型的CMT症状,而有的个体仅有轻微症状,甚至有的携带致病基因个体无症状[5]。
疾病诊断
一、神经电生理
电生理检查对于区分脱髓鞘性和轴索性神经病十分重要,同时可以检测是否有临床下的感觉神经受累,有助于CMT的分型;此外,节段性运动神经传导检测在脱髓鞘型CMT与CIDP的鉴别中也有重要作用。均匀的神经传导速度减慢(上肢运动神经传导速度<38m/s)提示脱髓鞘型CMT(CMT1以及CMT4),而神经传导速度正常或轻度减慢(正中或尺神经运动传导速度>38m/s)、伴有复合肌肉动作电位及感觉动作电位波幅降低提示CMT2。当上肢的运动神经传导速度位于25~45m/s的中间值时,需要警惕CMTX1。脱髓鞘型CMT的运动神经传导速度通常均匀减慢,若出现明显的波形离散、传导阻滞通常提示CIDP可能性大。但在MPZ基因突变的CMT1B中,偶尔会出现传导阻滞;CMTX1中,有时可有不对称的传导速度减慢,可有明显的波形离散甚至传导阻滞[8]。
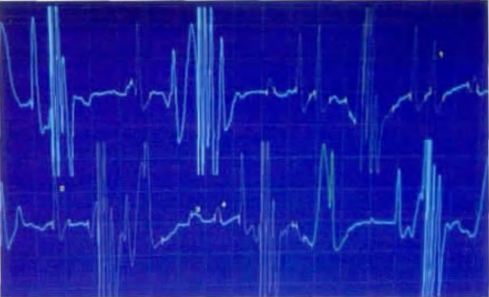
图6.CMT患者针极肌电图。表现为轻微收缩时可见运动单位时限增宽,波幅增高,多项波增强 [8]
二、基因检测
基因检测对于CMT的诊断和分型十分重要。鉴于PMP22、MPZ、GJB1及MFN2基因突变在CMT中占90%以上,故可以根据患者的临床和电生理特征选择可能相关的基因进行一代测序检测。随着高通量测序技术的普及,对于常染色体显性的脱髓鞘性周围神经病,可首先采用MLPA技术进行PMP22基因重复突变的检测,如果阴性再选择高通量测序方法对更多相关基因进行检测。
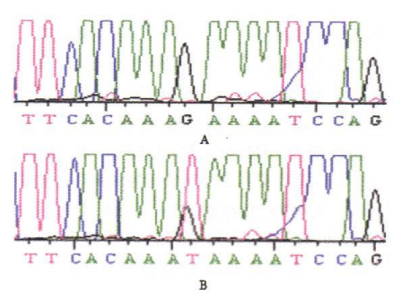
图7.基因测序图。A:正常对照;B:CMT患者[9]
三、腓肠神经活体组织检查
随着基因检测方法应用,绝大多数疑诊病例无需进行神经活检。但当临床及肌电图不典型时,可通过神经活检来协助鉴别诊断。取外踝和跟腱正中切口,取材后用4%多聚甲醛固定,石蜡包理,行HE染色及免疫组织化学检测。选择病变明显部位进行定位修块超薄切片,电镜观察[8]。
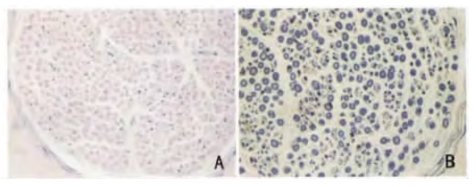
图8.CMT患者腓肠肌神经病理改变。A:HE染色;B:Weils染色。
A、B均可见大量神经纤维轴索变性,部分髓鞘肿胀。[8]
动物模型
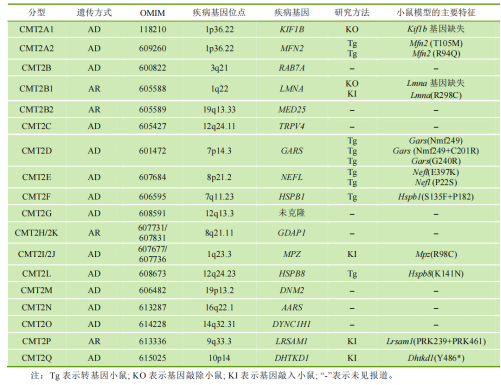
迄今已报道的CMT2致病基因共有17个,在已经报道的CMT2亚型中,有些亚型已建立了小鼠模型,并开展了小鼠的表型分析及机制研究;有些亚型正在构建小鼠模型,为后续研究做准备。由此产生相应的CMT2亚型,CMT2各亚型的遗传方式、OMIM编号、疾病基因名称及定位、小鼠模型的构建方法及主要特征等见表1[10]。
表2.CMT2亚型及其小鼠模型一览表[10]
治疗方式
目前尚无逆转腓骨肌萎缩症病程的治疗方法,主要是对症支持治疗,包括康复治疗、外科矫形手术、药物治疗等,以最大限度恢复独立活动能力、提高生活质量和尽可能减少残疾的发生与发展为目标。应根据患者年龄、骨骼畸形类型和程度、肌力失衡范围和程度,以及患者治疗期望值等因素制定治疗方案,此外,还应高度重视患者心理问题,上述综合治疗方案需多学科协作。随着越来越多致病基因的逐步明确和相关发病机制的深入认识,特异性靶向治疗成为腓骨肌萎缩症治疗的发展方向,目前的研究重点在于制定基于发病机制的治疗策略,并已开展相关药物基础与临床试验[11]。
一、传统的对症支持治疗
1.康复治疗
康复治疗在腓骨肌萎缩症疾病管理中占主导地位,以改善行走能力和生活质量为基本目标,包括力量训练和拉伸训练以维持肌力、防止肌萎缩,以及适当的辅具(矫形器)以鼓励患者活动并提高安全性,同时嘱患者控制体重,避免肥胖增加运动负担。
2.外科矫形手术
腓骨肌萎缩症患者足部畸形是逐步进展的过程,儿童期和青春期患儿表现为柔性高弓内翻足畸形,随着年龄增长进展为固定畸形。早期以穿戴矫形鞋联合物理治疗为主,尽量避免外科手术;而对于足踝畸形致功能障碍严重患者,可早期予外科手术;已形成固定畸形或畸形严重患者应采取积极的外科手术治疗。手术治疗原则是纠正足部畸形,重建和平衡足踝肌力。
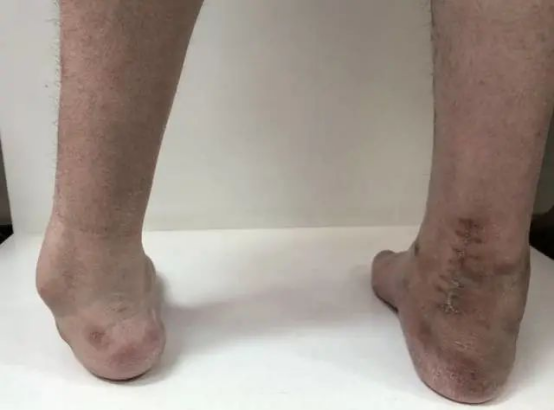
图9.CMT患者接受手术治疗[13]
3.其他方法
疼痛是腓骨肌萎缩症的常见症状,主要是神经性疼痛,包括痉挛和感觉异常;部分为骨关节疼痛,表现为背部、膝关节、踝关节、足部和手部疼痛。常用的神经病理性疼痛治疗药物如三环类抗抑郁药和抗惊厥药难以缓解疼痛,运动训练和物理治疗可以使疼痛减轻。
二、靶向治疗
表3.靶向治疗
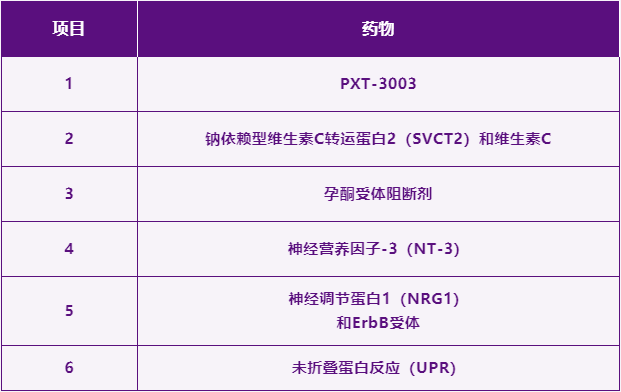
以上药物均处于临床试验阶段。
目前尚无根治腓骨肌萎缩症的药物,通过个体化康复训练和必要的外科矫形手术可以使患者运动功能和生活质量得以改善;二代基因测序(NGS)技术使分子诊断水平得到显著提高,基于基因诊断的遗传咨询可以有效减少新病例的发生;以直接作用于相关基因、蛋白质和调节网络为靶点,以修复周围神经系统蛋白表达异常为目标的疾病修饰疗法试验正在开展并有望取得新成果;腓骨肌萎缩症疾病测量工具尚待改进和开发,方可更好地用于临床病程的精确评价和新药临床试验的开展。
声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考文献及照片来源:
[1]孙顺昌,张海鸥.腓骨肌萎缩症研究概述[J].中国神经精神疾病杂志,2010,36(01):60-63.
[2]贺慧芬,王萌,郭爱红,张瑶,丁江博,李杰,刘越,王丙聚.腓骨肌萎缩症的治疗研究进展[J].临床医学研究与实践,2021,6(06):196-198.DOI:10.19347/j.cnki.2096-1413.202106068.
[3]郭鹏,翟晖,宋福聪.腓骨肌萎缩症临床表现、基因分型和分子发病机制研究进展[J].中风与神经疾病杂志,2013,30(10):953-955.DOI:10.19845/j.cnki.zfysjjbzz.2013.10.029.
[4]马迪. 腓骨肌萎缩症的新致病基因鉴定及功能研究[D].南方医科大学,2017.DOI:10.27003/d.cnki.gojyu.2017.000039.
[5]黄玉柱,李晓捷,周丽,王婷,朱琼.运动疗法和矫形器治疗对于腓骨肌萎缩症康复的研究进展[J].医学理论与实践,2020,33(06):891-893.DOI:10.19381/j.issn.1001-7585.2020.06.012.
[6]https://m.haodf.com/neirong/wenzhang/7767195108.html?os=ios
[7]https://m.youlai.cn/baike/disease/s5qhn55n7m.html
[8]羊毅,蒋军林,江泓,史峥莉.腓骨肌萎缩症电生理、病理和基因定位研究[J].国际神经病学神经外科学杂志,2014,41(04):317-319.DOI:10.16636/j.cnki.jinn.2014.04.011.
[9]张付峰,唐北沙,赵国华,罗巍,夏昆,刘小民,肖剑锋,张如旭,陈彪,张成,潘乾,蔡芳,郭鹏.腓骨肌萎缩症患者致病基因突变特点的研究[J].中华医学杂志,2005(26):1809-1812.
[10]于珍,栾春杰,顾鸣敏.腓骨肌萎缩症2型(CMT2)小鼠模型的研究进展[J].遗传,2014,36(01):21-29.
[11]辛颖.X-连锁肾上腺脑白质营养不良诊治[J].中国实用儿科杂志,2016,31(06):429-433.
[12]https://mp.weixin.qq.com/s/Gig4D4d0-ls8XQsTTjcNpQ
[13]https://mp.weixin.qq.com/s/wo_Jsq4X6lCnf4yYmZV6og