

脊髓性肌萎缩症(SMA)[3]——携带不同SMN2拷贝数的人源化小鼠模型
脊髓性肌萎缩症(Spinal Muscular Atrophy,SMA)是由SMN1基因突变引发的遗传性神经肌肉疾病,导致SMN蛋白缺乏,脊髓前角运动神经元退化,造成肌无力和肌萎缩。作为婴幼儿最常见的致死性遗传病,SMA发病越早病情越重,2岁以下儿童中位生存期仅约10个月[1]。在上两期疾病科普中(脊髓性肌萎缩症(SMA)[1]、脊髓性肌萎缩症(SMA)[2]),我们了解了SMA的基本信息、常用模型、诊疗指南等。今天,我们来继续了解人体内存在与SMN1高度同源的SMN2基因,虽产生的功能性SMN蛋白较少,但SMN2拷贝数越多,患者症状通常越轻。因此,调控SMN2表达已成为SMA治疗的重要方向,旨在增加功能性SMN蛋白来缓解症状[2]。
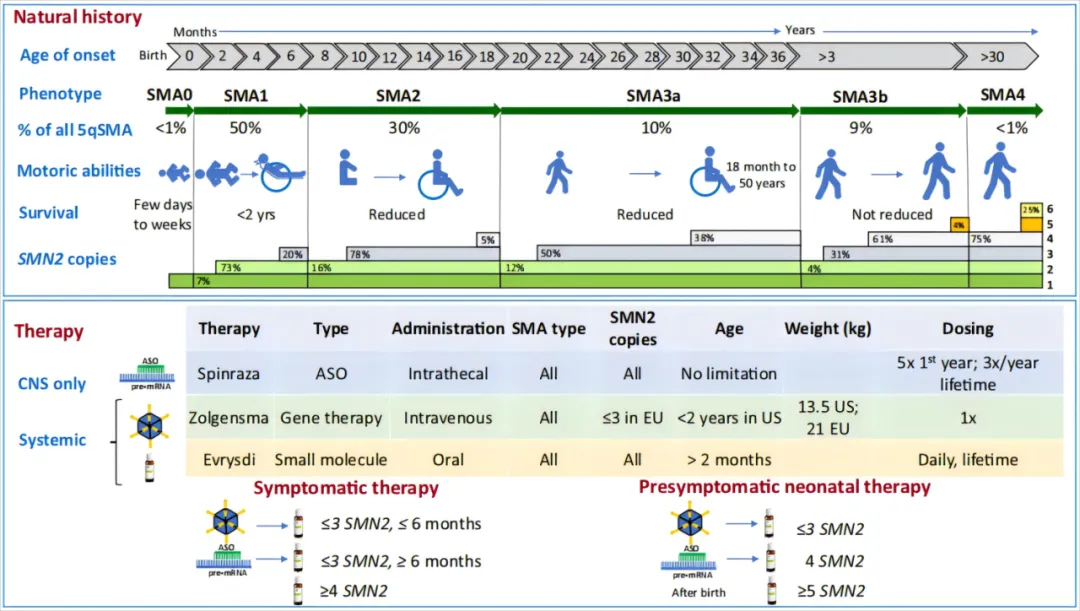
图1 SMA的疾病自然史及疗法[2]
一、致病机理
运动神经元存活基因1(SMN1)在人体全身广泛表达,尤其在脊髓中含量丰富。其编码的SMN蛋白被誉为细胞生存的“管家蛋白”,在剪接体蛋白复合体的组装中扮演关键角色,直接影响运动神经元的存活、功能及神经突起的发育[3-4]。当SMN1双拷贝缺失时,SMN蛋白的严重缺乏会导致脊髓运动神经元RNA剪接异常和功能障碍,引发神经元退化和肌肉失去神经支配,最终导致肌无力和萎缩,极大威胁患者的运动能力和生命[3-4]。因此,早期干预至关重要——在神经元不可逆丢失前启动治疗,才能有效挽救生命并促进运动功能的恢复。
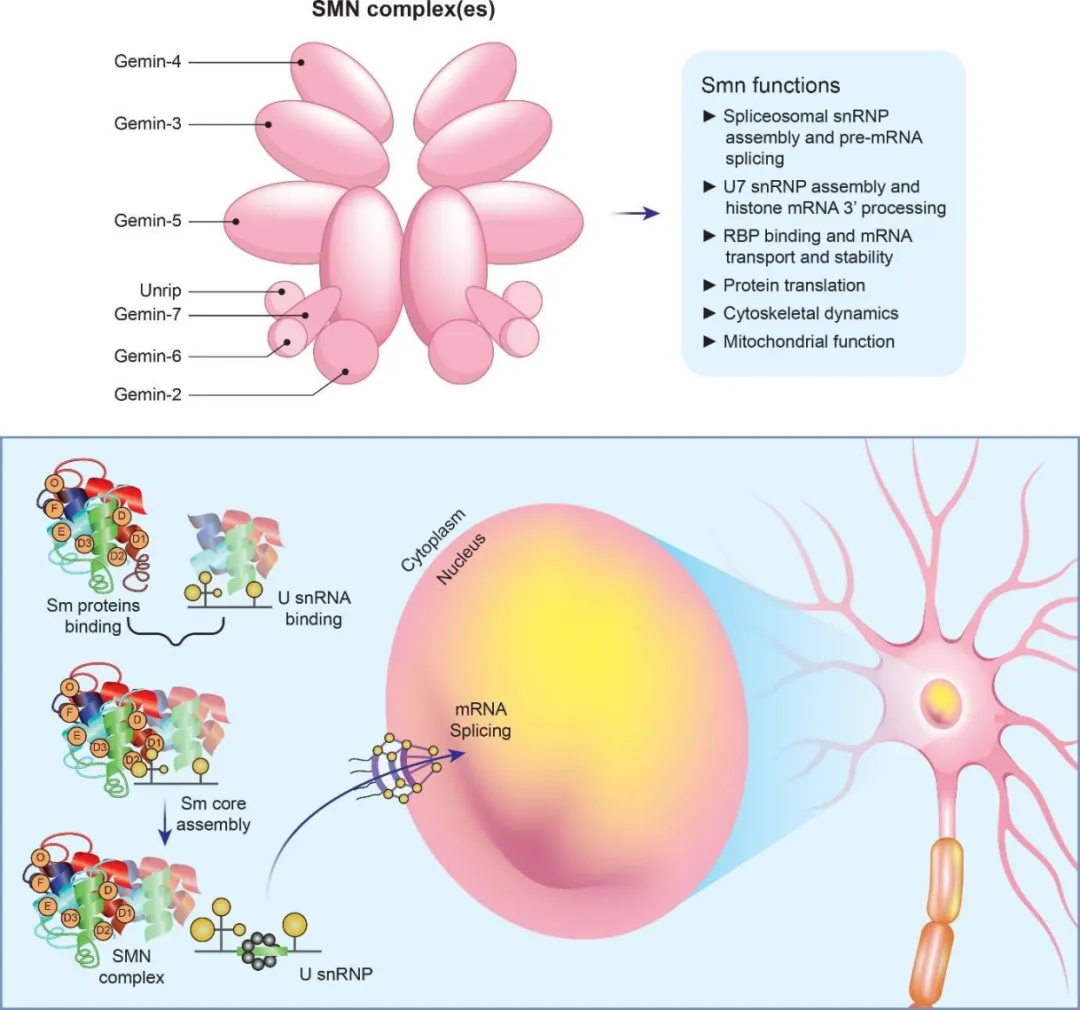
图2 SMN蛋白对于剪接体snRNPs组装至关重要,其缺失会影响mRNA剪接和表达[5]
二、SMN2基因拷贝
除了SMN1,人体内还存在与其高度同源的SMN2基因,二者仅在少数核苷酸上有所不同[6]。然而,SMN2基因在第7号外显子剪接增强子处存在c.840C>T的点突变,破坏了剪接增强子或形成剪接抑制子,导致大部分通过SMN2前体mRNA剪接生成的成熟mRNA缺失第7号外显子,生成的截短SMN蛋白功能丧失且迅速降解[7]。仅约10%的SMN2前体mRNA能够生成全长、功能性SMN蛋白[8]。在约95%的SMA患者中,SMN1双拷贝缺失使得SMN2成为功能性SMN蛋白的唯一来源,但其产量不足以完全代偿SMN1缺失,导致疾病发生[6-8]。
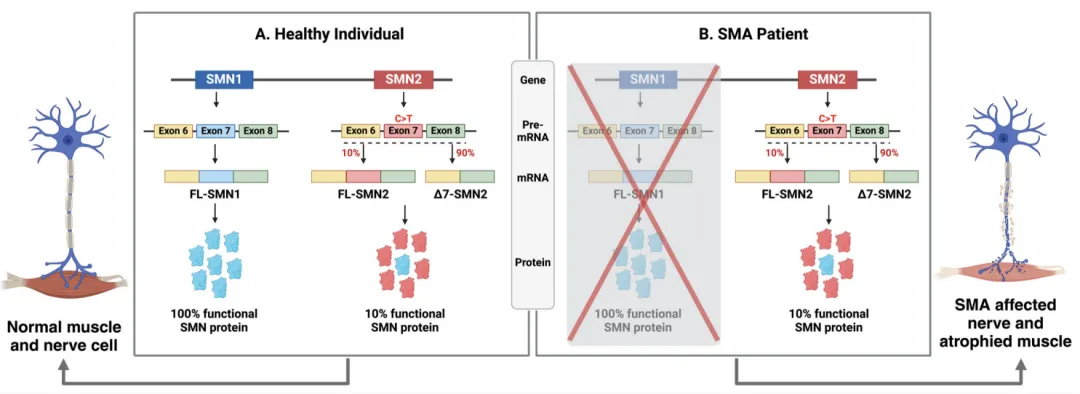
图3 第7号外显子剪接增强子处的差异致使SMN2生成的功能性SMN蛋白远低于SMN1[9]
研究表明,SMN2拷贝数是SMA严重程度的关键“调节器”。患者体内SMN2拷贝数通常在1至6个之间,拷贝数越高,全长SMN蛋白越多,临床症状往往越轻[10-11]。
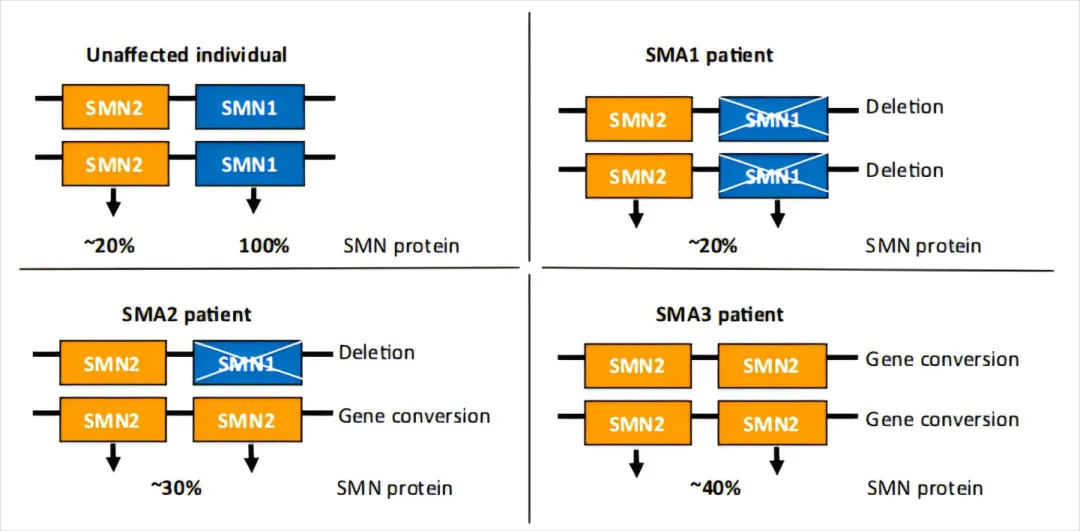
图4 SMN2拷贝数与SMA严重程度(Ⅰ型、Ⅱ型、Ⅲ型)之间的负相关关系[2]
基于SMN2的潜力,调控其剪接或表达以增加功能性SMN蛋白已成为SMA治疗的热点方向。目前获批的三款SMA药物中,两款靶向SMN2:Biogen的反义寡核苷酸药物Nusinersen和罗氏的口服剪接调节剂Risdiplam,均通过调节SMN2剪接增加功能性蛋白的生成[12]。2024年,这两款药物的销售额分别达到15.7亿美元和近18亿美元,凸显了其临床价值和市场潜力[12]。
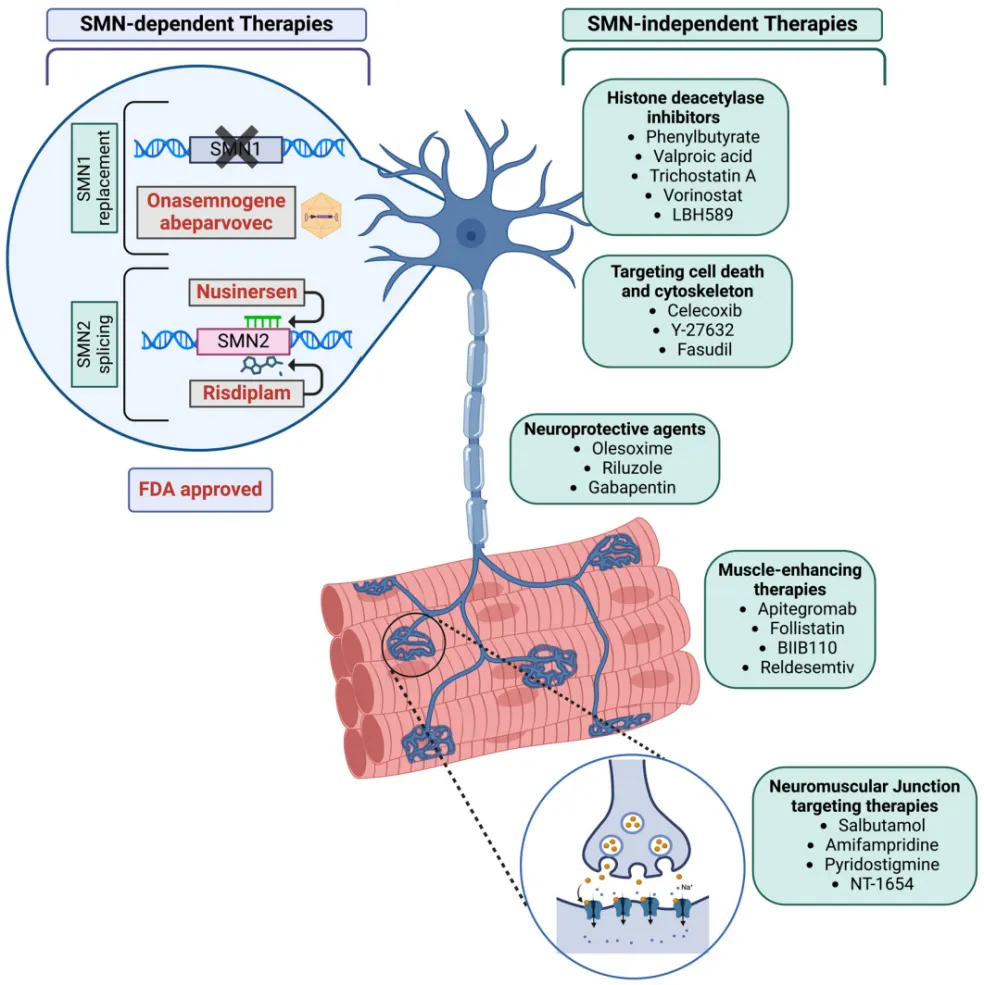
图5 靶向调控SMN2 mRNA剪接模式是SMN依赖性疗法的主要方式[9]
三、小鼠模型
与人类不同,小鼠仅拥有单一SMN基因(Smn1),其敲除会导致胚胎致死,难以直接模拟SMA的致病机制及SMN2的补偿作用[13]。构建既能反映SMA致病机制(尤其是SMN2拷贝数的影响)又符合人类病程发展的人源化小鼠模型,对于开发和验证SMN2靶向疗法具有重要意义。联盟单位赛业生物采用两种策略构建了SMN2人源化基础品系,并通过繁育获得携带不同SMN2拷贝数的SMA小鼠模型。
● Smn1hSMN2/hSMN2小鼠:将小鼠Smn1双拷贝基因原位替换为人类SMN2双拷贝,模拟携带2拷贝SMN2的SMA患者;
●ROSA26hSMN2/hSMN2小鼠:在小鼠ROSA26安全位点上插入双拷贝人源SMN2基因。
通过多代繁育,最终获得Smn1缺失背景下携带2、3和4拷贝SMN2的SMA小鼠模型,分别为B6-2*hSMN2小鼠(产品编号:C001504)、B6-3*hSMN2小鼠(产品编号:C001681)和B6-4*hSMN2小鼠(产品编号:C001682)。
*B6-hSMN2:https://www.cyagen.cn/mice-products/C001504
*B6-3*hSMN2:https://www.cyagen.cn/mice-products/C001681
*B6-4*hSMN2:https://www.cyagen.cn/mice-products/C001682
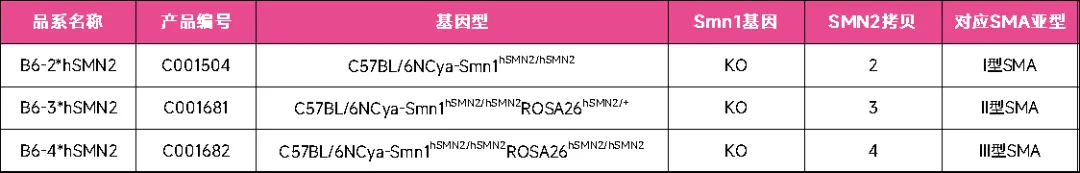
综上,B6-2*hSMN2(产品编号:C001504)、B6-3*hSMN2(产品编号:C001681)和B6-4*hSMN2(产品编号:C001682)小鼠在完全缺失鼠源Smn1基因表达的情况下,分别携带2、3和4拷贝人源SMN2基因,分别模拟人类Ⅰ型、Ⅱ型和Ⅲ型SMA的遗传特性。数据表明,这些小鼠在全长SMN2转录本及功能性SMN蛋白表达、生长发育和生存情况等方面,与携带相应SMN2拷贝数的人类SMA患者病程大致匹配。因此,此类人源化小鼠模型不仅能较好地模拟SMA病因,特别是SMN2拷贝数对疾病进展的影响,同时为研究SMA发病机制及开发针对人源SMN2的治疗策略提供了宝贵工具。
四、参考文献
[1]Mercuri E, Sumner CJ, Muntoni F, Darras BT, Finkel RS. Spinal muscular atrophy. Nat Rev Dis Primers. 2022 Aug 4;8(1):52.
[2]Wirth B. Spinal Muscular Atrophy: In the Challenge Lies a Solution. Trends Neurosci. 2021 Apr;44(4):306-322.
[3]Wirth B, Karakaya M, Kye MJ, Mendoza-Ferreira N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu Rev Genomics Hum Genet. 2020 Aug 31;21:231-261.
[4]Nicolau S, Waldrop MA, Connolly AM, Mendell JR. Spinal Muscular Atrophy. Semin Pediatr Neurol. 2021 Apr;37:100878.
[5]Meneri M, Abati E, Gagliardi D, Faravelli I, Parente V, Ratti A, Verde F, Ticozzi N, Comi GP, Ottoboni L, Corti S. Identification of Novel Biomarkers of Spinal Muscular Atrophy and Therapeutic Response by Proteomic and Metabolomic Profiling of Human Biological Fluid Samples. Biomedicines. 2023 Apr 23;11(5):1254.
[6]Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin. 2015 Nov;33(4):831-46.
[7]Day JW, Howell K, Place A, Long K, Rossello J, Kertesz N, Nomikos G. Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatr. 2022 Nov 3;22(1):632.
[8]Butchbach MER. Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. Int J Mol Sci. 2021 Jul 23;22(15):7896.
[9]Haque US, Yokota T. Recent Progress in Gene-Targeting Therapies for Spinal Muscular Atrophy: Promises and Challenges. Genes (Basel). 2024 Jul 30;15(8):999.
[10]Cuscó I, Bernal S, Blasco-Pérez L, Calucho M, Alias L, Fuentes-Prior P, Tizzano EF. Practical guidelines to manage discordant situations of SMN2 copy number in patients with spinal muscular atrophy. Neurol Genet. 2020 Nov 18;6(6):e530.
[11]Calucho M, Bernal S, Alías L, March F, Venceslá A, Rodríguez-Álvarez FJ, Aller E, Fernández RM, Borrego S, Millán JM, Hernández-Chico C, Cuscó I, Fuentes-Prior P, Tizzano EF. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018 Mar;28(3):208-215.
[12]FiercePharma. (2025, February 12). Roche nabs FDA nod: Evrysdi tablets gaining potential convenience edge over SMA meds Biogen. Retrieved March 20, 2025, from https://www.fiercepharma.com/pharma/roche-nabs-fda-nod-evrysdi-tablets-gaining-potential-convenience-edge-over-sma-meds-biogen
[13]Edens BM, Ajroud-Driss S, Ma L, Ma YC. Molecular mechanisms and animal models of spinal muscular atrophy. Biochim Biophys Acta. 2015 Apr;1852(4):685-92.
来源:赛业生物
【声明】本文为转载文章,本平台仅作分享、传递信息,版权归原作者所有,如有侵权,请联系删除。