

四氢生物喋呤缺乏症(BH4D)——PKU的“孪生姊妹”
疾病概述
通过之前每周知识关于苯丙酮尿症(PKU)的科普文章,相信大家对该疾病已有初步了解。其实它还有一个“孪生姊妹”,那就是四氢生物喋呤缺乏症(tetrahydrobiopterin deficiency,BH4D)。
四氢生物喋呤(tetrahydrobiopterin,BH4)是一种芳香族氨基酸羟化酶的辅助因子,其合成或代谢途径中酶的先天性缺陷会是导致BH4D的罪魁祸首,造成神经递质合成受影响,出现高苯丙氨酸血症(HPA)以及严重的神经系统损害症状和智能障碍。BH4D是HPA的一种亚型,是一种常染色体隐形遗传病,与PKU同属于HPA[1]。
各国的BH4D在HPA中的比例不一,马来西亚较高占64%。截至2015年,BH4D在中国大陆的南方地区发病率高于北方,南方BH4D约占HPA的29%,北方约占6%~7%,中部地区约占14%[4]。

图1.BH4D患者主要症状 图片来源于网络
致病机理
BH4D既往又称恶性PKU,该病是因四氢生物喋呤代谢途径中任何一种合成酶或还原酶缺乏,导致四氢生物喋呤生成不足甚至完全缺乏所致。
BH4缺乏一方面影响苯丙氨酸羟化酶的稳定性,阻碍苯丙氨酸代谢,进而导致血中苯丙氨酸浓度增高,出现类似苯丙酮尿症的症状;另一方面由于降低了苯丙氨酸羟化酶、酪氨酸羟化酶及色氨酸羟化酶的活性,导致神经递质前体左旋多巴胺和5-羟色胺生成受阻,影响脑内神经递质(多巴胺、5-羟色胺)的合成,使患者出现严重的神经系统损害症状和体征,因此其临床症状比苯丙酮尿症更严重,预后更差[2]。
疾病分类
迄今已知有5种酶参与BH4的代谢[3]
合成BH4需要3种酶:
鸟苷三磷酸环化水解酶(GTP cyclohydrolase,GTPCH)
Ⅰ型‚6-丙酮酰四氢喋呤合成酶(6-pyruvoy-l tetrahydropterin synthase,PTPS)
墨喋呤还原酶(sepiapterinreductase,SR)
再生BH4需要2种酶:
二氢喋啶还原酶(dihydropteridine reductase,DHPR)
喋呤-4α-甲醇氨脱水酶(pterin-4α-carbinolamine dehydratase,PCD)
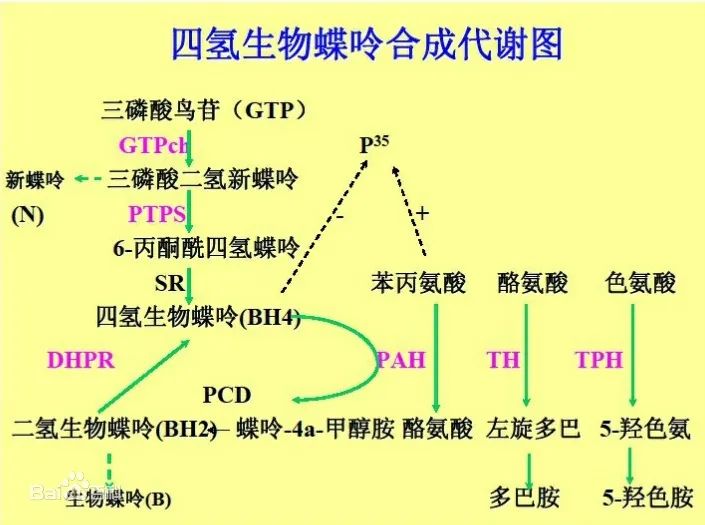
图2.四氢生物喋呤合成代谢图[4]
至今尚未发现SR缺乏导致的BH4缺乏,故现在常将BH4缺乏症分成以下4种类型:PTPS缺乏症、DHPR缺乏症、GTPCHⅠ型缺乏症、PCD缺乏症。
由于PTPS缺乏症的临床表现具有高度异质性,故又分为3种类型:经典型(严重型)、部分型(外周型)、暂时型。
临床表现
BH4缺乏早期除血苯丙氨酸(Phe)增高外无任何临床症状,故往往被误认为非经典型PKU,给予低(无)Phe奶粉治疗,一些患儿经治疗后血Phe很快下降至正常或接近正常,但会出现进行性神经系统损害症状,往往在出生后3个月逐渐出现以下症状:
多巴胺缺乏症状,如运动障碍、肌张力低下、嗜睡、眼震颤、吸吮力低下、吞咽困难等;
5-羟色胺缺乏相关症状,如反应迟钝、抑郁、失眠等;
去甲肾上腺素缺乏相关症状,如躯干肌张力低下、四肢肌张力增高、眼睑下垂、小脑发育障碍等;
还会有顽固性抽搐、严重小头畸形,不明原因的高热,运动里程碑发育迟滞,婴儿或儿童期仍然不能独坐、站、行走,全身瘫痪,智能发育严重障碍等[5]。
图3.BH4D缺乏症患者 图片来源于:北京康旭医学
其中PTPS的三种类型临床表现各不相同:
严重型患者PTPS完全缺乏,脑脊液中神经递质代谢产物水平下降,表现严重的神经系统症状;
部分型或外周型患者PTPS轻度缺乏,脑脊液中神经递质代谢产物水平大多正常,患者仅表现为高苯丙氨酸血症,无其他神经系统症状;
暂时型为PTPS成熟延迟所致,随着酶的完全成熟,临床表现逐渐消失。
辅助检查[6]
1.血苯丙氨酸测定
(1)荧光定量法:检测干血滤纸片中苯丙氨酸(Phe)浓度,正常血Phe浓度<120μmol/L(2mg/dl),血Phe浓度>120μmol/L提示高苯丙氨酸血症(HPA)。
(2)串联质谱法:检测干血滤纸片中 Phe 及酪氨酸(Tyrosine,Tyr)浓度,并可自动计算Phe与Tyr比值(Phe/Tyr)。血Phe浓度>120μmol/L及Phe/Tyr>2.0提示为HPA。
2.尿喋呤谱分析:是目前国内鉴别诊断苯丙酮尿症(PKU)及各型四氢生物喋呤缺乏症(BH4D)的重要方法。采用高效液相色谱分析法,测定新喋呤(neopterin,N)、生物喋呤(biopterin,B)浓度,并计算生物喋呤比例B%[B/(B+N)×100%]。各种酶缺乏患儿呈现不同的尿喋呤谱,见表1。
表1.不同病因导致的HPA生化特点[6]
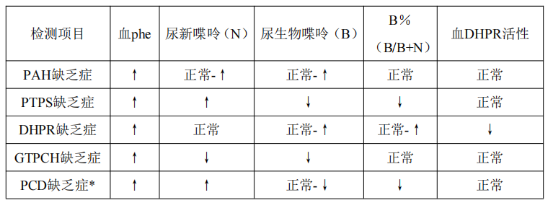
注:↑.增高;↓.降低;*.尿中出现7-生物喋呤
3.红细胞DHPR活性测定:是二氢喋啶还原酶(DHPR)缺乏症的确诊方法。需采用双光束分光光度计测定干滤纸血片中红细胞DHPR活性。DHPR缺乏症患儿DHPR活性显著降低。
4.BH4负荷试验:为BH4缺乏症的辅助诊断方法及BH4反应性PKU/HPA的判断方法,需在留取尿喋呤标本后进行。对于轻度HPA或已用特殊饮食治疗后血Phe浓度已降低者,可先做尿喋呤分析及DHPR活性测定,对诊断不确定者再进行BH4负荷试验。试验前及试验过程中正常饮食[3]。
5.基因诊断:是HPA病因的确诊方法,建议常规进行。尤其对经上述鉴别诊断试验仍不能明确诊断者更需尽早进行基因诊断。至今已报道BH4缺乏症相关基因变异:
编码PTPS酶的基因PTS位于11q22.3-q23.3,包含6个外显子,东亚地区已发现107种PTS基因突变类型。中国PTS基因热点突变为c.155A>G、c.259C>T、c.286G>A和c.IVSl-291A>G(占76.9%),c.155A>G、c.259C>T、c.286G>A导致严重型PTPS缺乏症,c.166G>A及c.IVSl-291A>G变异可能与轻型PTPS缺乏症有关。
DHPR基因QDPR位于4p15.3,含7个外显子,已报道66种基因突变类型。
其他少见的与BH4缺乏症相关的基因包括PCBD1、GCH1、SPR和DNAJC12基因。
6.头颅影像学检查:有助于评价患儿脑损伤的程度。
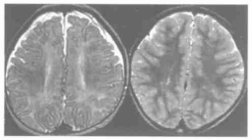
图4.23周BH4D患儿,左图T2WI示额、枕叶髓鞘发育延迟。右图为治疗1年后该患儿的髓鞘发育情况,显示髓鞘发育达到正常同龄儿的水平[2]
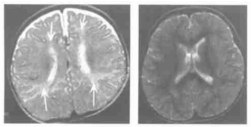
图5.24周BH4D患儿,左图T2WI示双侧侧脑室前后角和体部可见异常高信号。右图为该患儿治疗1年后T2WI上异常高信号消失[2]
7.脑电图检查:对合并癫痫患者应进行脑电图检查。
疾病诊断
1.诊断
BH4D患儿早期除血Phe增高外(血Phe浓度持续>120mmol/L及Phe/Tyr>2.0),无明显临床表现,易被误诊为PAH缺乏所致PKU。给予低(无)Phe奶粉治疗后,患儿血Phe浓度虽很快下降,但神经系统损害症状却逐渐出现,主要表现为躯干肌张力低下,四肢肌张力增高等。因此对所有诊断高苯丙氨酸者,应及时进行尿喋呤谱分析(在低Phe饮食治疗前)、DHPR活性测定,或联合BH4负荷试验来进行鉴别诊断,早期确诊BH4D,并结合基因测序分析判断属于哪一种BH4D。
(1)PTPS缺乏症:出生体重多偏低,尿新喋呤(N)明显增加,生物喋呤(B)明显降低,B%<10%(多<5%),BH4负荷试验其血Phe浓度在服用4~6小时下降至正常,PTS基因检测到变异。
(2)DHPR缺乏症:尿生物喋呤多明显增高,红细胞DHPR活性极低,QDPR基因检查到变异。
(3)GTPCH缺乏症:尿新喋呤、生物喋呤均极低,B%正常。但常染色体显性遗传性GTPCH缺乏所致多巴反应性肌张力低下症(DRD)主要表现为多巴胺递质缺乏,而无5-羟色胺递质缺乏症状及高苯丙氨酸血症,以一侧或双侧肢体运动障碍,逐步影响至其他肢体,晨起或休息后症状好转,呈昼间波动现象。苯丙氨酸负荷试验显示苯丙氨酸羟化酶活性降低导致负荷试验后血苯丙氨酸下降缓慢,尿喋呤谱分析可显示新喋呤、生物喋呤偏低。单纯多巴治疗效果显著。GCH1基因检测到变异。
2.确诊标准
(1)临床表现:在PKU临床表现的基础上,并有肌力与肌张力低下等。
(2)血Phe浓度>120μmol/L及Phe/Tyr>2.0。
(3)尿喋呤谱异常,或血DHPR活性异常。
(4)BH4负荷试验,结果符合BH4缺乏症的特征。
(5)检测到BH4D基因致病性变异。若只检测到一个BH4D相关基因,但符合上面(1),(2),(3),(4)项者可诊断。
治疗方式
1.综合治疗
心理指导:BH4D是需要终生治疗的疾病,需提高治疗依从性,达到良好的疗效。入学后需要告知学校老师,配合饮食及教育指导,做好患儿的心理辅导工作。
2.特异性治疗
经新生儿筛查诊断的患儿多无临床症状,难以判断严重型与轻型。诊断明确后可按不同病因尽早给予BH4或无Phe特殊饮食及神经递质前体治疗,提倡终生治疗。
(1)BH4或特殊饮食治疗:目的是降低血Phe浓度。PTPS缺乏症、GTPCH缺乏症及PCD缺乏症患者在正常饮食下,补充BH4[1~5mg/(kg·d)],分2次口服,使血Phe控制到正常水平;DHPR缺乏症及BH4治疗困难的患儿采用低Phe特殊奶粉或饮食治疗(同PKU治疗),使血Phe浓度控制到接近正常水平(120~240μmol/L)。
(2)神经递质前体等治疗:绝大多数PTPS缺乏症及DHPR缺乏症都需要神经递质前体多巴(左旋多巴)及5-羟色氨酸联合治疗[1];轻型PTPS缺乏症可不服用神经递质前体。左旋多巴、5-羟色氨酸宜从1mg/(kg·d)开始,每周递增1mg/(kg·d)(表2)。有条件时可根据脑脊液神经递质代谢产物水平或临床表现调节药物治疗剂量。血清泌乳素可作为多巴剂量调节的参考指标,多巴剂量不足也可导致泌乳素浓度增高。此外,DHPR缺乏症患儿易合并继发性脑叶酸缺乏症,需补充四氢叶酸(亚叶酸钙)5~20mg/d。
表2.各年龄段患儿神经递质前体治疗量mg/(kg·d)[1]

(3)基因治疗的基础研究:各型BH4缺乏症均由基因突变导致酶活性下降或丧失引起,基因治疗是最彻底的治疗方法。此外,BH4具有广泛的功能,口服BH4和神经递质前体并不能完全代替BH4的生理功能,因此,目前疗法的效果并非尽如人意。
基因治疗目前还仅限于实验研究,应用于临床还有很长的距离。PTPS缺乏症的基因治疗研究较多。早在1996年有学者以逆转录病毒为载体,将PTPS cDNA导入原代培养的PTPS缺乏症患者的皮肤成纤维细胞。以细胞因子刺激这些细胞,发现合成BH4的能力有所恢复,但这种方法合成的BH4的量较少。后来人们发现,正常成纤维细胞不表达GPTCHⅠ型基因,所以PTPS缺乏症患者的皮肤成纤维细胞既缺乏PTPS,也缺乏GTPCHⅠ型酶。将GTPCHⅠ型和PTPS的cDNA均导入原代培养的PTPS缺乏症患者的皮肤成纤维细胞,这种成纤维细胞合成BH4的能力较强,可释放200pmol~800pmol/106cell/day的BH4。如将这种细胞植入不同的器官,包括中枢神经系统,可作为移植细胞释放BH4[3]。
声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考文献及图片来源:
[1]蔡宗友,胡定波,曹小英,陈秀銮,王伟婷,黄海.四氢生物喋呤缺乏症的新生儿筛查和诊断治疗[J].中国优生与遗传杂志,2016,24(08):82+128.DOI:10.13404/j.cnki.cjbhh.2016.08.036.
[2]张知新,喻唯民,王琳,周忠蜀,沈抒,张雪哲.四氢生物喋呤缺乏症治疗前后神经系统表现及其脑白质病变分析[J].临床儿科杂志,2005(02):101-104.
[3]高宏,喻唯民.四氢生物喋呤缺乏症的研究进展[J].中日友好医院学报,2001(05):295-298.
[4]https://baike.baidu.com/item/%E5%9B%9B%E6%B0%A2%E7%94%9F%E7%89%A9%E8%9D%B6%E5%91%A4/5607741?fr=aladdin
[5]杨蓉,王枫,徐小兰,黄志华.新生儿苯丙酮尿症与四氢生物喋呤缺乏症筛查与治疗[J].江西医药,2005(10):25-27.
[6]https://www.nrdrs.org.cn/app/rare/disease-list-article.html