

先天性肌无力综合征(CMS)——睁不开眼的“瞌睡”宝宝
疾病概述
当你的宝宝出现眼睑下垂,睁不开眼,易疲劳,肌无力的症状一定要提高警惕啦,快带着宝宝去医院检查,他很有可能患上先天性肌无力综合征。
先天性肌无力综合征(congenital myasthenic syndrome,CMS)是由美国梅奥诊所的Engel教授于1977年首次报道,该患者是1例出生后不久即出现活动后力弱、易疲劳、腱反射减低、重复电刺激波幅递减以及抗胆碱酯酶药物无反应的患儿,电镜发现运动轴索的数量及突触后膜皱褶密度明显降低,生化分析发现终板特异性的乙酰胆碱酯酶明显减少,推测该患儿为先天性肌无力综合征。

图1.CMS患者主要症状 图片来源于网络
CMS是由于参与神经肌肉接头(neuromu scular junction,NMJ)的形成、结构及其维持修饰相关的蛋白,以及神经递质传递相关信号蛋白存在先天性缺陷,从而导致神经末梢至运动终板信号传递功能障碍的一组遗传异质性疾病,临床上主要表现为肌无力和易疲劳。CMS是一种可部分治疗的罕见病,英国的患病率约为3.8/100万,部分罕见亚型在世界范围内仅有数例报道[1]。
致病机理
CMS发病机制为负责神经肌肉信号传递的蛋白质功能异常,不同基因变异导致的CMS,其发病年龄、临床表现、电生理特点及治疗策略亦有不同。由于近年来人们对CMS认识的不断深入以及高通量测序技术的普及,目前已有30多个基因被证实与CMS相关(图2)[2]。

图2.神经肌肉接头结构及已知的CMS相关蛋白分布示意图[2]
疾病分类
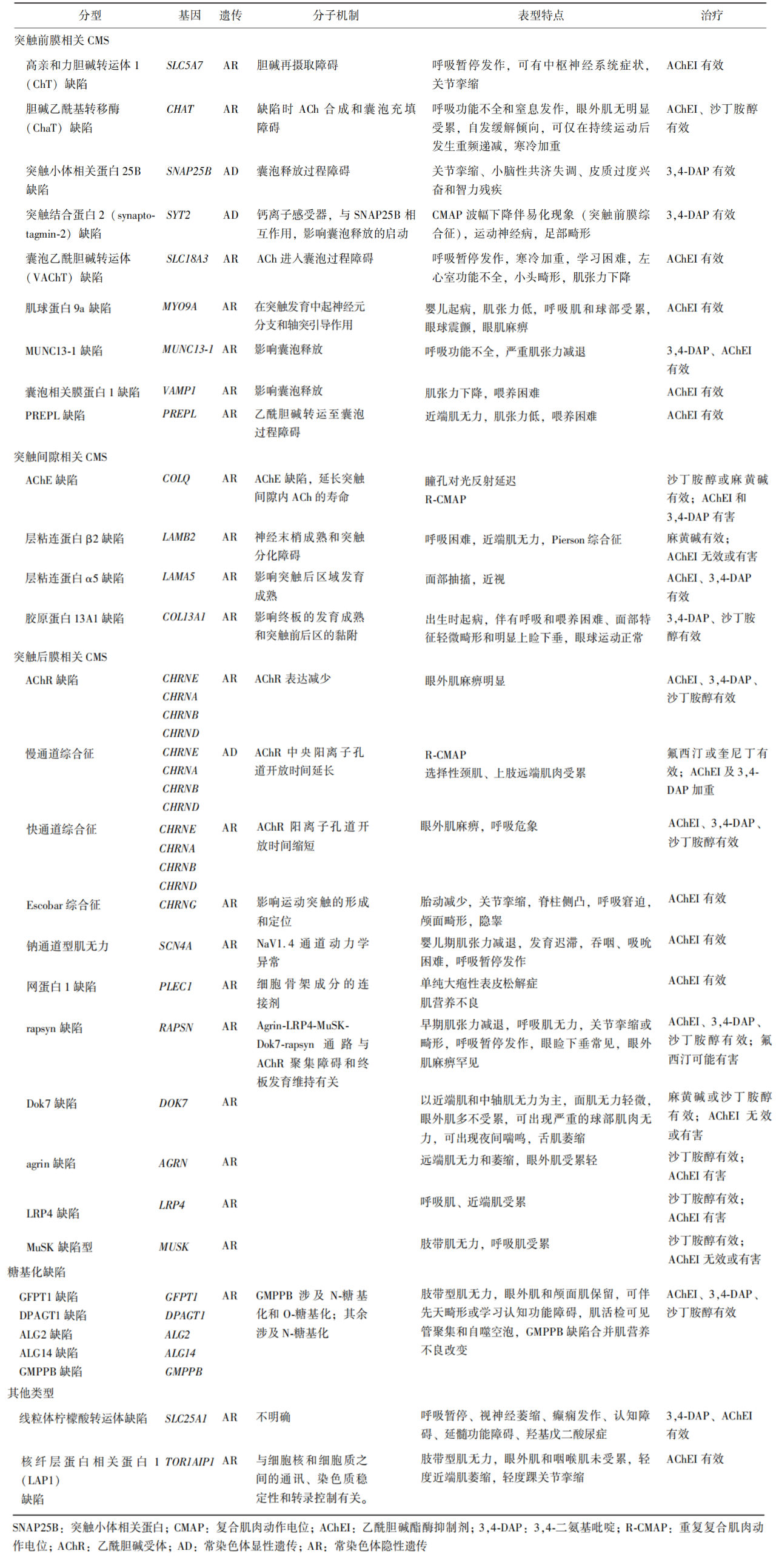
人们通常依据缺陷基因所编码蛋白在NMJ的解剖学分布和功能,将CMS分为以下5种类型:突触前膜蛋白缺陷、突触间隙蛋白缺陷、突触后膜蛋白缺陷、糖基化缺陷和其他罕见类型(表1)[2]。
表1.CMS亚型的分子机制、表型特点和药物选择[2]
临床表现
CMS具有一些共同的临床表现[3]:
(1)CMS起病早,常在出生后或婴幼儿期发病,缓慢进展。
(2)临床主要表现为眼部、躯干、肢体肌肉力量弱。常有喂养困难、哭声低、眼睑下垂、吞咽呛咳和运动发育迟滞等症状。
(3)不耐受疲劳,症状可能在发热、感染等诱因下突然加重。
(4)心肌和平滑肌通常不受累。因为遗传机制的不同,各CMS亚型可具有相对特异的临床表现(详情见上表,表1)。

图3.CMS患者眼睑下垂(图片来源于网络)
辅助检查[1]
1.血清学检查
肌酸激酶正常或轻度升高;抗AChR抗体、抗MuSK抗体、抗AChE抗体等抗体均阴性,这是诊断CMS的必要条件。
2.神经电生理检查
RNS示波幅递减10%以上。一般情况下,低频(2~3Hz)RNS即可出现波幅递减,部分未受累的肌肉在静息状态下也可不出现波幅递减改变,但以10Hz频率长时间(5~10min)刺激骨骼肌仍可诱发出显著波幅递减现象。SFEMG示异常颤抖或阻滞。运动神经传导检测中单个神经刺激出现重复复合肌肉动作电位(CMAP)是慢通道综合征、终板AChE缺陷CMS的特征性表现,也可见于服用大剂量胆碱酯酶抑制剂患者。

图4.ColQ综合征可见R-CMAP波[4]
注:A.刺激尺神经后,出现 CMAP 波;B.刺激面神经后,主峰呈双峰且出现CMAP波;
C.正常对照,刺激尺神经后,无CMAP波出现
3.对胆碱酯酶抑制剂反应性
可通过静脉注射依酚氯铵或肌内注射甲基硫酸新斯的明或口服胆碱酯酶抑制剂对照观察试验进行评估。对儿童来说,一般选择甲基硫酸新斯的明试验。对于仅有疲劳性肌无力而无眼睑下垂、球肌受累表现的患者,也可通过口服胆碱酯酶抑制剂对照观察试验进行评估。
4.基因检测
当怀疑一种特定的临床综合征时,进行Sanger测序可快速精确锁定致病基因的突变。同时,当锁定一种特定的致病基因时,推荐进行Sanger测序进行家系验证。当临床表现不典型时,可以选择高通量测序方法对多种可能相关的基因进行检测筛查。采用全外显子测序有可能发现未知的基因突变,但其致病性需要验证,强调临床和电生理资料对诊断是十分重要的[5]。
疾病诊断
依据临床特征及辅助检查,可做出临床诊断,确诊需要进行基因检测。当临床研究指向某个/某些候选基因时(表2),对特定CMS的遗传诊断有很大帮助,可选择单基因测序或靶向捕获二代测序[1]。
表2.先天性肌无力综合征基因诊断线索[1]

注:CMAP:复合肌肉动作电位;RNS:重复神经电刺激;EPP:终板电位
CMAP:compound muscle action potential;RNS:repetitive electrical nerve stimulation;
EPP:end-plate potentia
动物模型
研究员构建了最常见的Dok7CM, Dok71124-1127dup以及Dok7 Y396F,Y406F小鼠,研究DOK7缺失C端区域如何导致神经肌肉突触结构和功能异常,发现与WT小鼠相比,DOK7CM小鼠出生时纯合小鼠数量极少,膈肌中突触分化前后突触数量降低,且突触不成熟;DOK7(2YF)成年鼠具生育能力,神经肌肉突触数量基本正常,但DOK7CM小鼠中MUSK活性降低(图5),推断MUSK活性降低导致Dok7CM的疾病[6]。

图5.在 Dok7CM/CM小鼠中CRK向突触和MUSK DOK7复合物的募集受损[6]
治疗方式
CMS的治疗药物主要有胆碱酯酶抑制剂、钾离子通道阻滞剂、AChR阻滞剂和β2受体激动剂,免疫抑制剂治疗无效。溴吡斯的明和3,4-DAP在数小时内发挥作用,而奎尼丁、氟西汀、麻黄碱和沙丁胺醇作用较慢,可能在数天、数周或数月内发挥作用[2-3]。
1.胆碱酯酶抑制剂
溴吡斯的明,通过抑制AChE分解乙酰胆碱而发挥作用。溴吡斯的明是CMS的一线治疗药物,除了CHRNA1、CHRNB1、CHRND、CHRNE突变导致的慢通道CMS,以及COLQ、LAMB2、DOK7、MUSK、AGRN、LRP4、PLEC基因突变有关的CMS外,其他亚型的CMS均首选溴吡斯的明治疗,其中,慢通道CMS、COLQ、DOK7、LRP4、LAMB2亚型禁用溴吡斯的明。
2.钾离子通道阻滞剂
3,4-DAP阻断突触前膜快电压门控钾离子通道,延长突触前膜去极化,从而增加动作电位持续时间和乙酰胆碱释放。3,4-DAP是CMS的二线治疗药物,多作为溴吡斯的明的添加治疗,也在一些对胆碱酯酶抑制剂无反应如MUSK、PLEC突变中,可能有效。
3.AChR阻滞剂
氟西汀、奎尼丁在慢通道综合征中作为长效通道阻滞剂缩短AChR通道开放时间,阻止突触后膜去极化块的形成和受体在生理条件下的脱敏。由于奎尼丁更容易引起心脏传导障碍,干扰依赖细胞色素P450ⅡDA通路的药物,在实践中氟西汀更为常用。然而,由于氟西汀有产生自杀意念不良反应的危险,儿童和青少年,必须在精神科监测下使用,大剂量可延长QT间期。
4.β2受体激动剂
麻黄碱和沙丁胺醇对CMS的治疗作用机制不明。推测它们可稳定NMJ并降低AChR的分散。β2受体激动剂是COLQ、DOK7、MUSK、AGRN、LRP4突变的一线治疗药物,也在一些胆碱酯酶抑制剂治疗有效但剂量需求大的患者中作为添加治疗发挥作用,如原发性AChR缺陷、Rapsyn缺陷、糖基化缺陷CMS。
5.其他治疗
对症支持治疗:包括理疗、言语治疗;矫形器、步行器或轮椅;呼吸支持等。
声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考文献及照片来源:
[1]刘志梅,沈新明,方方.先天性肌无力综合征诊治进展[J].中华实用儿科临床杂志,2021,36(11):876-880.DOI:10.3760/cma.j.cn101070-20201105-01723.
[2]王文卿,赵玉英,焉传祝.先天性肌无力综合征[J].罕见病研究,2022,1(02):110-121.
[3]肖婷,吴丽文.先天性肌无力综合征的诊治进展[J].中国当代儿科杂志,2020,22(06):672-677.
[4]戴毅等.终板胆碱酯酶缺失型先天性肌无力综合征一家系的临床、电生理、肌肉病理和基因研究. 中华神经科杂志,2016,49(8):604-609.
[5]https://www.nrdrs.org.cn/app/rare/disease-list-article.html
[6]https://wap.sciencenet.cn/blog-3479614-1308183.html