

RDDC邀您关注大疱性表皮松解症:皮肤的求救信号
疾病概述
大疱性表皮松解症(Hereditary Epidermolysis Bullosa, EB),亦称半桥粒大疱性表皮松解症或遗传性大疱性表皮松解症,构成了一组复杂的机械性大疱性皮肤疾病谱系。其核心临床特征是皮肤及黏膜在遭遇轻微外伤或摩擦后,迅速形成水疱,如图1所示。这一病理过程根源于编码表皮角蛋白或真表皮间锚定蛋白的基因缺陷,进而引发皮肤结构的根本性改变,表现为皮肤脆性显著增加及创伤后水疱的易发。在组织病理学层面,EB呈现为表皮内或表皮下水疱形成,且通常不伴随炎症细胞浸润。依据皮肤分离层次的差异,EB被细分为单纯型、交界型及营养不良型三大类。
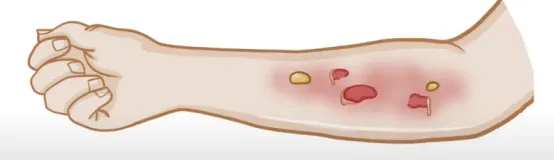
图1. EB皮肤表征
来源:Osmosis from Elsevier
在大疱性表皮松解症(EB)的多种表现形式中,相较于轻度类型,其严重形式的皮肤外表现更为普遍。尽管如此,值得注意的是,特定亚型的EB往往伴随着独特的疾病并发症。如图2所示:具体而言,交界性EB的患者可能罹患间质性肺病及肾病综合征,这些并发症显著增加了疾病的复杂性。另一方面,单纯型EB的患者则可能面临幽门闭锁的风险,这进一步凸显了疾病对消化系统的影响。
营养不良性大疱性表皮松解症(Dystrophic Epidermolysis Bullosa, DEB),它尤为显著地表现为小口畸形、强直性咬合以及假性并指等特征,这些临床表现深刻反映了该疾病过程中反复发生的水泡形成、炎症反应、瘢痕增生以及皮肤纤维化的广泛性与深度。这些复杂的病理变化不仅影响了患者的外貌与生活质量,也对治疗提出了更高的挑战。
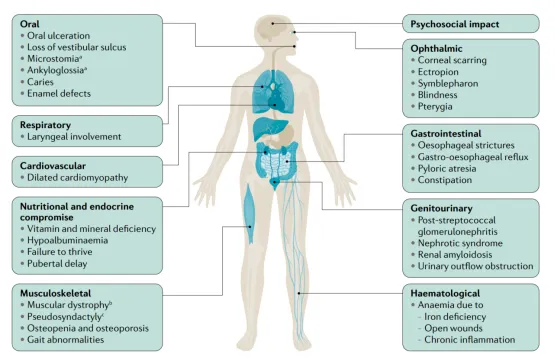
图2. EB的皮肤外表征[1]
病因和流行病学
DEB疾病依据其遗传特性的差异,被更细致地划分为常染色体显性遗传型DEB(Dominant Dystrophic Epidermolysis Bullosa, DDEB)与隐性遗传型DEB(Recessive Dystrophic Epidermolysis Bullosa, RDEB)。这两类DEB均根源于COL7A1基因中的一个或多个基因突变[2],这一点通过RDDC数据库的深入剖析(如图3所示)得到了明确,揭示了COL7A1作为DEB的唯一关键关联基因,迄今已记录有511种与之相关的突变形式。
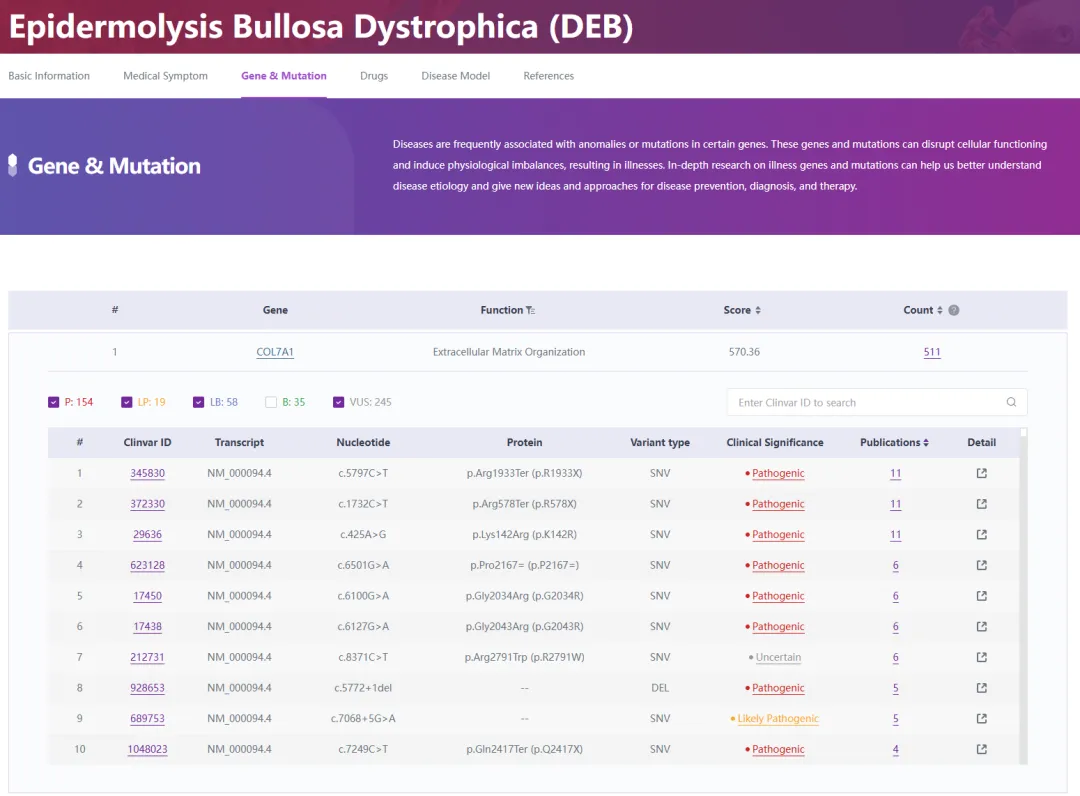
图3. DEB与COL7A1基因的关系
来源:RDDC罕见病数据中心
了解更多疾病基因信息,欢迎登陆RDDC官网(https://rddc.tsinghua-gd.org),开启您的罕见病研究AI之旅。
COL7A1基因的主要功能是编码VII型胶原蛋白(COL7),这种蛋白质在构建连接真皮(皮肤深层结构)与表皮(皮肤外层)的锚定纤维中发挥着不可或缺的作用。由于DEB患者体内缺乏功能正常的锚定纤维,他们的皮肤变得异常脆弱,即便是轻微的摩擦或创伤也足以引发水疱和撕裂,严重影响生活质量。
进一步而言,DEB患者面临的开放性伤口极易导致皮肤感染,长期的炎症反应可能促使皮肤纤维化,进而引发手指和脚趾的粘连(并指/并趾),并显著增加罹患侵袭性鳞状细胞癌的风险,这是一种潜在致命的皮肤恶性肿瘤。因此,DEB不仅对患者的生活造成极大困扰,还构成了严重的健康威胁。
尽管EB在全球范围内的总发病率相对较低,约为1/53,000,但DEB作为其中最为严重的类型之一,对患者的生命质量和日常生活构成了严峻挑战。该疾病无性别倾向,可发生于任何年龄段及种族,且其遗传特性决定了家族中病例的聚集现象[3]。
症状及诊断
DEB的症状表现极具标志性,患者往往在出生或出生后不久即展现出皮肤的极度脆弱。轻微的物理接触,如衣物摩擦、拥抱或轻触,均可能迅速触发水疱的形成。这些水疱多见于四肢伸侧、手掌、足底等易摩擦区域,且愈合过程缓慢,常伴随显著的瘢痕遗留。随着病情进展,患者可能面临皮肤增厚、萎缩、钙化乃至关节挛缩(如爪形手)等严重后果。此外,DEB还可能累及消化系统(如吞咽困难)、眼部(如角膜瘢痕、视力损害)及牙齿等系统,造成广泛影响。
确诊DEB需综合考量多方面信息。确诊需综合家族史、临床表现、组织病理学、透射电镜及基因诊断技术,后者能精确识别COL7A1基因突变,明确遗传类型。
模型
为了研究DEB的发病机制、疾病进程以及潜在的治疗方法,科学家开发了多种动物模型,其中最常用的是Col7a1基因敲除小鼠模型[4]。这种模型通过基因编辑技术敲除小鼠体内的Col7a1基因,使其表现出与人类DEB相似的皮肤脆弱性症状,是研究DEB发病机制和治疗方法的理想模型。同时,小鼠模型具有与人类相似的生理结构和基因组,能够较好地模拟人类疾病的发生和发展过程。
基因治疗
目前,DEB的传统治疗主要侧重于支持护理和伤口处理,以防止水泡的形成和控制症状。然而,这些方法并不能根治DEB,只能在一定程度上缓解患者的痛苦。基因治疗作为一种新兴的治疗方法,为DEB患者带来了新的希望。基因治疗旨在通过修复或替换导致疾病的缺陷基因,从而恢复患者的正常生理功能。基因治疗通过向患者体内递送正常的COL7A1基因或其功能性拷贝,以恢复VII型胶原蛋白的表达,从而改善患者的皮肤状况。
根据智慧芽数据库的DEB基因治疗药物研发情况可知(图4),目前仅有Beremagene geperpavec-svdt(B-VEC)进入上市阶段, 除此之外,还有多家机构正在研发针对DEB的基因治疗药物,这些药物大多处于临床前或临床试验阶段。这些机构包括中国的无锡科金生物科技有限公司、北京博雅汇康生物科技有限公司等,以及国外的Stanford University、University of Southern California等。这些在研药物的研究方向可能包括不同的基因递送载体、治疗策略和优化方案等。
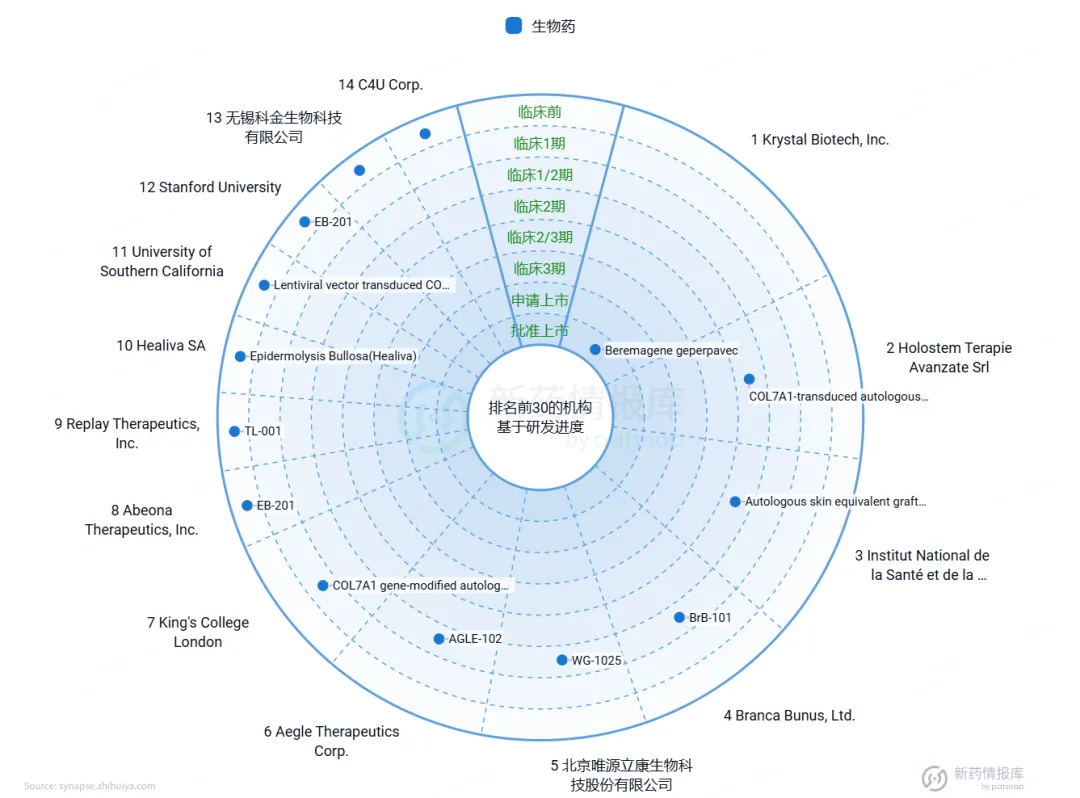
图4. DEB的基因治疗药物研发现状
来源:智慧芽新药情报库
B-VEC该药物采用基于非整合、复制缺陷的单纯疱疹病毒1型 (HSV-1)载体编码两个全长人类 COL7A1 拷贝,在局部给药至 DEB 伤口后恢复 COL7蛋白(图5)。B-VEC于2023年在美国获批,成为首个局部基因治疗药物,也是首个获批的DEB治疗药物[3]。B-VEC是一种非侵入性、局部应用、可重复给药的基因疗法,采用凝胶剂型设计,由皮肤科医生或初级护理医师直接应用于患者皮肤DEB伤口处,促进伤口愈合。B-VEC旨在将COL7A1基因的2个拷贝递送至DEB伤口的皮肤细胞中,制造出功能性COL7蛋白,从而在分子水平上治疗DEB,解决根本性的致病机制。
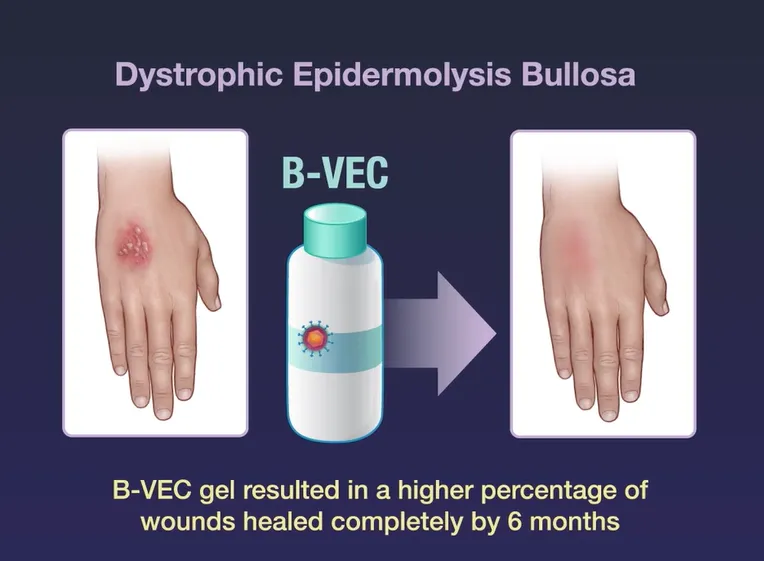
图5. B-VEC的基因治疗策略[5]
随着基因治疗技术的不断进步和临床数据的积累,未来有望看到更多针对DEB的基因治疗药物上市。这些药物将为患者提供更多的治疗选择,并有望显著改善患者的生活质量和预后。
RDDC助力罕见病研究
罕见病数据中心(Rare Disease Data Center,简称“RDDC”),是由联盟内成员单位合作开发的关于罕见病研究的数据库。RDDC整合了全球开源的流行病学、药物研发、疾病相关基因图谱、基因突变位点、大小鼠实验动物模型等数据信息,再结合人工智能、生物信息等先进技术,部署了致病性预测工具(Pathogenicity Predictor)、RNA剪接预测模型(RNA Splicer)、通路分析(Pathway Analysis)等一系列AI、生信工具,赋能罕见病诊疗研究工作。
欢迎感兴趣的用户复制链接(https://rddc.tsinghua-gd.org)或扫描下方二维码,体验RDDC罕见病数据中心。

扫码进入RDDC

声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考资料:
[1]A. Bardhan, L. Bruckner-Tuderman, I.L.C. Chapple, J.D. Fine, N. Harper, C. Has, T.M. Magin, M.P. Marinkovich, J.F. Marshall, J.A. McGrath, J.E. Mellerio, R. Polson, A.H. Heagerty, Epidermolysis bullosa, Nat Rev Dis Primers 6(1) (2020) 78.
[2]N. Dang, D.F. Murrell, Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa, Exp Dermatol 17(7) (2008) 553-68.
[3]K. Kridin, R.J. Ludwig, The Growing Incidence of Bullous Pemphigoid: Overview and Potential Explanations, Front Med (Lausanne) 5 (2018) 220.
[4]S. Takaki, T. Shimbo, K. Ikegami, T. Kitayama, Y. Yamamoto, S. Yamazaki, S. Mori, K. Tamai, Generation of a recessive dystrophic epidermolysis bullosa mouse model with patient-derived compound heterozygous mutations, Lab Invest 102(6) (2022) 574-580.
[5]S.V. Guide, M.E. Gonzalez, I.S. Bağcı, B. Agostini, H. Chen, G. Feeney, M. Steimer, B. Kapadia, K. Sridhar, L. Quesada Sanchez, F. Gonzalez, M. Van Ligten, T.J. Parry, S. Chitra, L.A. Kammerman, S. Krishnan, M.P. Marinkovich, Trial of Beremagene Geperpavec (B-VEC) for Dystrophic Epidermolysis Bullosa, N Engl J Med 387(24) (2022) 2211-2219.