

RDDC邀您关注Rett综合征:“美人鱼”女孩
疾病概述
Rett综合征(Rett Syndrome),又称雷特综合征,是一种严重影响儿童精神运动发育的罕见疾病。由于男性患者几乎无法存活,故Rett综合征好发于女性幼童。在出生后的6至18个月内,患病儿童会经历一段正常的神经和身体发展期,此后,Rett综合征的典型症状开始在儿童早期显现,并经历以下发展阶段:早发期、退化期、平台期及晚期运动退化(图1)[1,2]。
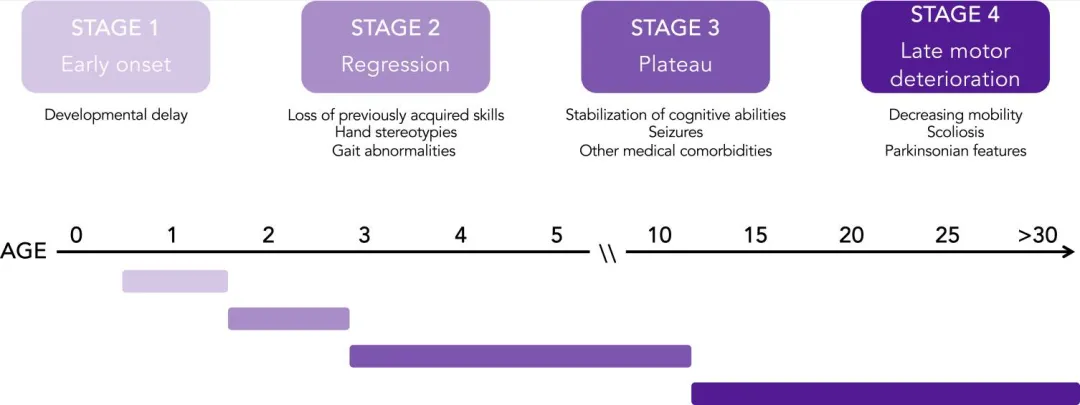
图1. Rett综合征患者的分期和症状发作时间表[2]
该综合征的特征性症状包括言语和运动技能的获得性丧失、重复的手部运动、呼吸不规则以及癫痫发作。此外,患者可能面临如散发性胃肠道问题、成长发育迟缓、早发性骨质疏松症、磨牙症以及突发性的尖叫等健康挑战。根据安安雷特罕见病研究所的患者症状统计(图2),大约35%的患者会表现出哭闹行为,19%有自伤行为,30%的患者出现无故尖叫,14%有伤害他人的倾向,18%的患者呈现脑电图异常,而9%的患者确诊为癫痫。
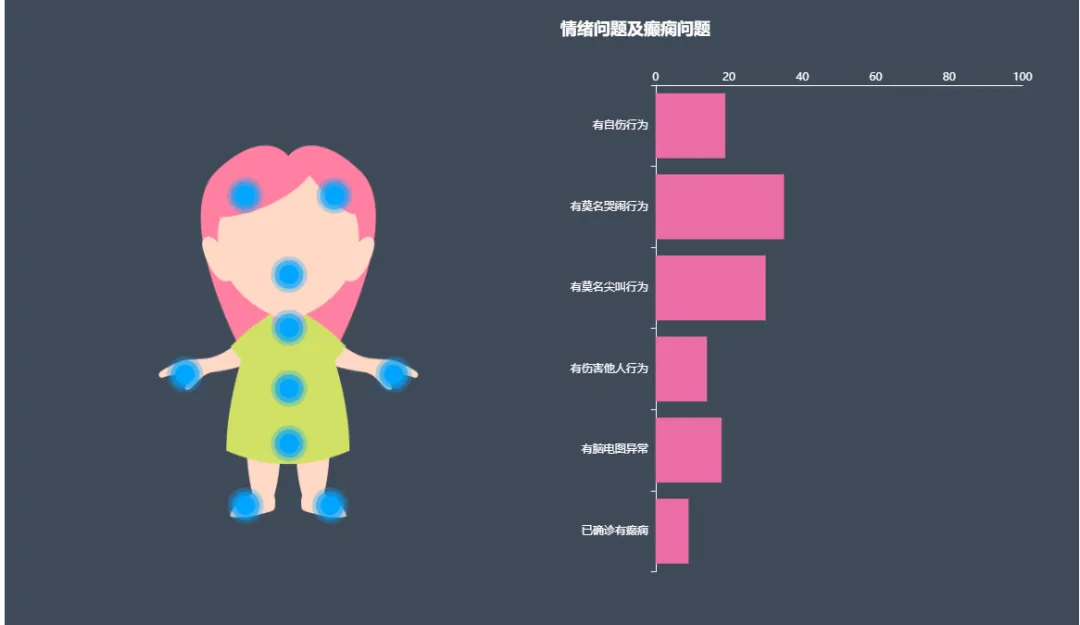
图2. Rett综合征患者症状统计
来源:安安雷特罕见病研究
作为一种罕见的遗传性脑部疾病,Rett综合征的影响遍及所有种族,女性发病率约为1/10,000[3]。从图3中我们可以清晰地看到,尽管中国Rett综合征患者的分布相对广泛,但主要集中在中东部地区,而西部地区由于地域辽阔、人口相对稀疏,患者分布较少。更为严峻的是,从医院分布的角度来看,总体数量较少,且分布严重不均,西部地区尤为缺乏相应的医疗资源。
因此,我们迫切需要加强对Rett综合征的认知,提高诊断率,并优化医疗资源配置,以改善患者的治疗条件和生活质量。这不仅需要医疗界的努力,也需要社会各界的关注和支持,共同为Rett综合征患者创造一个更加美好的未来。
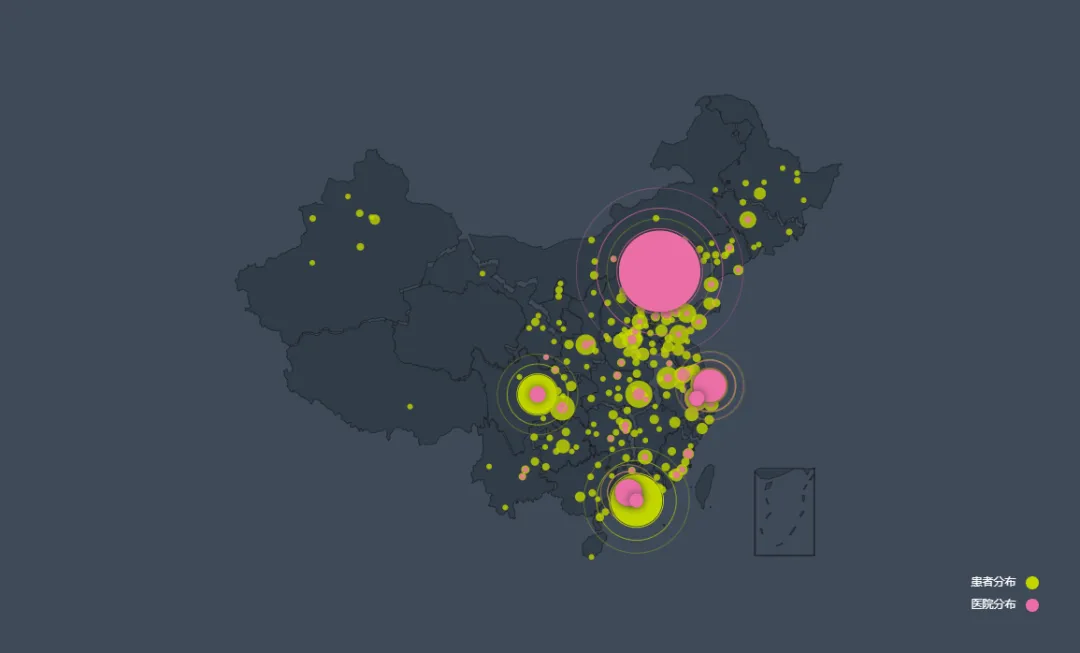
图3. Rett综合征患者及医院分布
来源:安安雷特罕见病研究所
相关基因及突变
在大约90%的患者中,Rett综合征的主要原因是MECP2基因的突变[4, 5]。MECP2,即甲基CpG结合蛋白2(Methyl-CpG Binding Protein 2),是一种多功能的转录调节因子,广泛存在于人体的各个细胞中,包括大脑(图4)。在脑组织中,MECP2基因对多种细胞类型,特别是神经元,具有关键作用,主要涉及维护神经元间的连接和细胞间通讯等关键功能[6]。
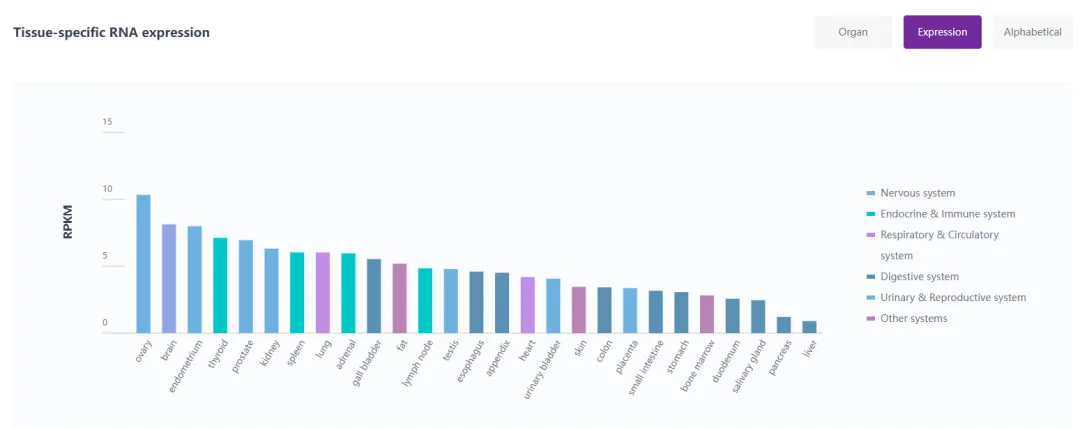
图4. MECP2基因组织特异性表达
来源:RDDC数据库
基因根据人类基因突变数据库,MECP2基因中已发现555个与Rett综合征相关的突变[7]。MECP2突变的广谱包括点突变、插入、重复、小或大缺失或整个MECP2基因缺失。通过RDDC数据库的检索(图4),我们了解到导致Rett综合征的致病突变高达389个,这些突变正成为医学研究的热点,并且相关的研究报道也在不断增多。这些研究对于深入理解Rett综合征的发病机理、改进诊断技术,以及开发潜在的治疗方法,都具有极其重要的价值。
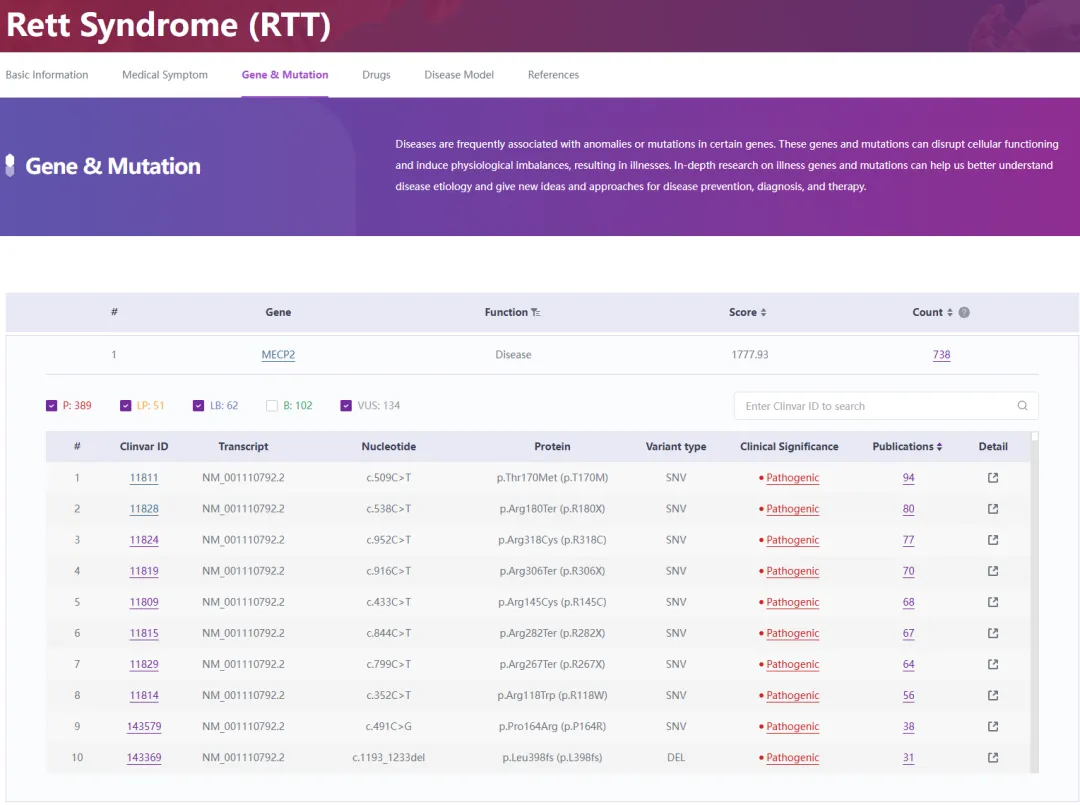
图5. MECP2基因突变与Rett综合征
来源:RDDC数据库
了解更多血友病基因信息,欢迎登陆RDDC官网,即可免费查阅!
疾病模型
Rett综合征的第一个小鼠模型是在2000年代初期开发的,来自 Adrian Bird和Rudolf Jaenisch的实验室,这两种模型都模拟了MECP2基因3号外显子的缺失。这些小鼠在生命初期的前4-5周内表现正常,但随后展现出一致且独特的行为表型,包括后肢张开的不寻常步态、尾巴悬吊时后肢紧握、毛发和胡须的杂乱状态、呼吸困难、震颤以及癫痫发作。这些行为特征逐渐加剧,并伴随一系列神经学症状,这些症状源于感觉运动或认知功能的异常,如焦虑相关行为的增加、与新生小鼠的社会互动减少,以及学习和记忆功能的缺陷。随着症状的出现,体重显著减轻,驼背和呼吸不规律现象变得更为严重,直至这些小鼠在出生后约10周时离世[8,9]。在随后开发的多种针对MECP2突变的多种Rett综合征模型小鼠表征[10]来看(图6),这些模型所展现的表型特征与Rett综合征高度一致,为深入研究该疾病的发病机制和潜在治疗方法提供了宝贵的工具。
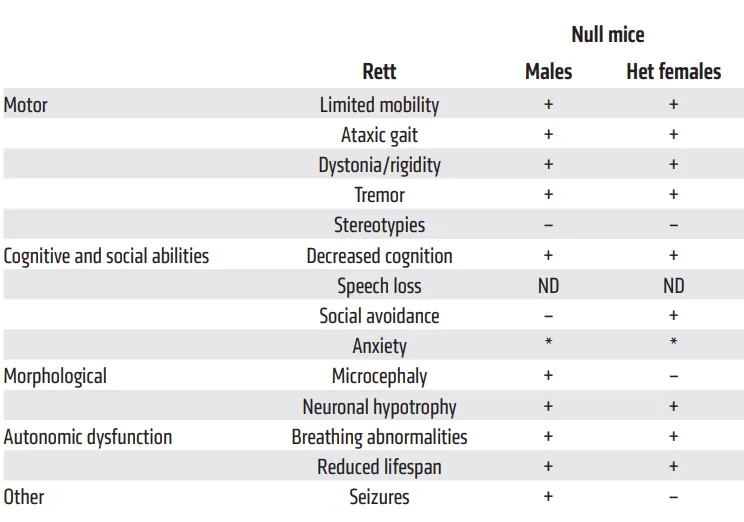
图6. Mecp2-/-与Rett表征对应[10]
治疗现状及前景
迄今为止,Rett综合征仍是一种无法根治的疾病,其治疗手段主要集中在缓解显著的临床症状并努力提升患者的生活质量。在寻求突破性的治疗方法中,基因治疗展现出极大的潜力(图7),并以其多样化的形式吸引了广泛关注。这些疗法包括但不限于通过基因替换技术来引入新的遗传物质,以弥补突变蛋白的缺失;或者利用基因组或RNA编辑技术,直接修改受Rett综合征影响的生物体的基因组或转录组,以期达到治疗疾病的目的[11]。
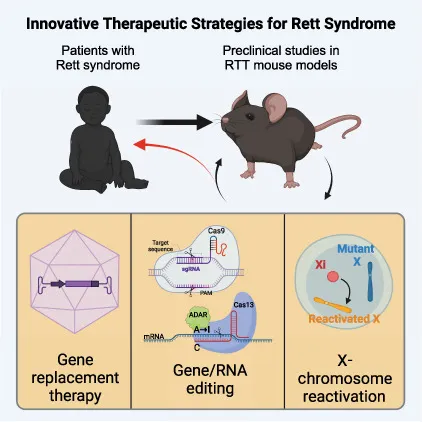
图7. Rett综合征的基因治疗策略[11]
根据我们从智慧芽新药情报库获取的数据(图8),针对Rett综合征的基因治疗药物研发正取得积极进展。目前共有7个不同的药物处于研发阶段,其中Neurogene公司的NGN-401已经进入临床1/2期试验,显示出令人期待的治疗前景。同时,在我国,上海金珂博生物技术公司也通过采用腺相关病毒技术,研发了一款名为Self-complementary adeno-associated virus 9 containing human methyl-CpG binding protein 2(简称Genecombio)的药物,目前正处于临床前试验阶段。这些进展共同为Rett综合征的治疗带来了新的希望。
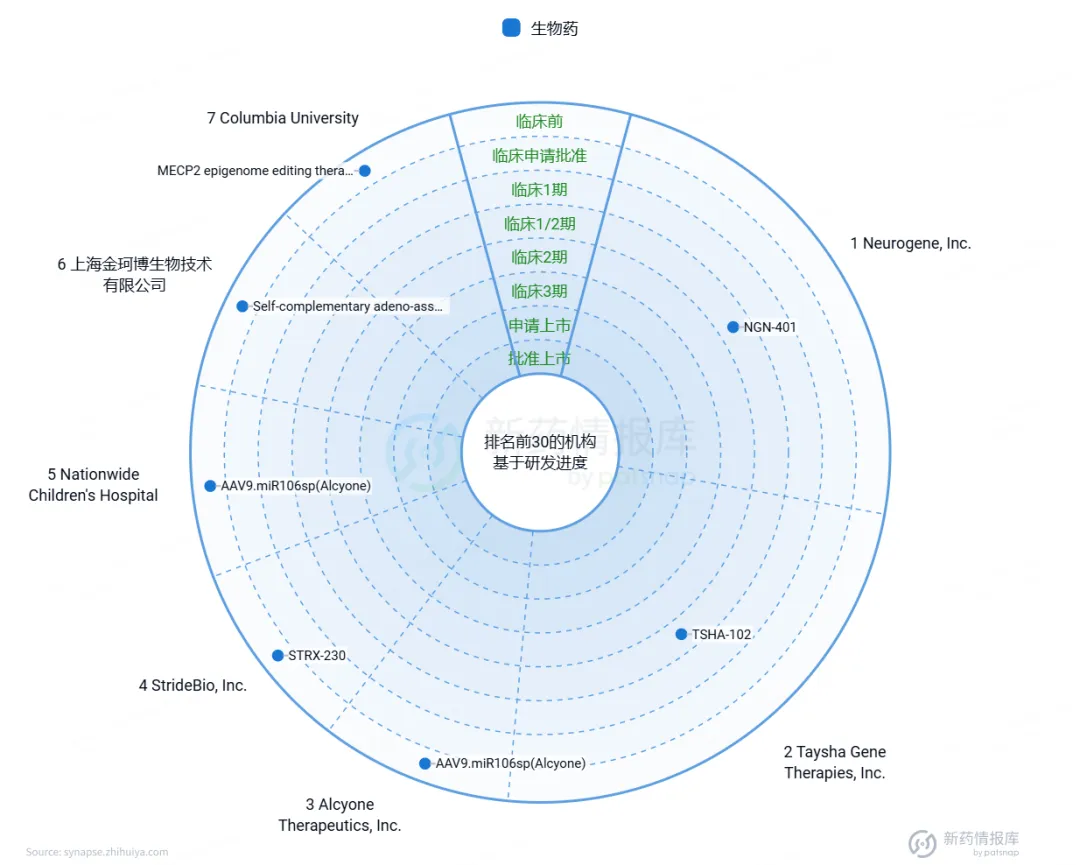
图8. Rett综合征基因治疗药物研发现状
来源:智慧芽新药情报库
RDDC助力罕见病研究
罕见病数据中心(Rare Disease Data Center,简称“RDDC”),是由联盟内成员单位合作开发的关于罕见病研究的数据库。RDDC整合了全球开源的流行病学、药物研发、疾病相关基因图谱、基因突变位点、大小鼠实验动物模型等数据信息,再结合人工智能、生物信息等先进技术,部署了致病性预测工具(Pathogenicity Predictor)、RNA剪接预测模型(RNA Splicer)、通路分析(Pathway Analysis)等一系列AI、生信工具,赋能罕见病诊疗研究工作。
欢迎感兴趣的用户复制链接(https://rddc.tsinghua-gd.org)体验RDDC网站。

声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考资料:
[1]S.M. Kyle, N. Vashi, M.J. Justice, Rett syndrome: a neurological disorder with metabolic components, Open Biol 8(2) (2018).
[2]B.E. Collins, J.L. Neul, Rett Syndrome and MECP2 Duplication Syndrome: Disorders of MeCP2 Dosage, Neuropsychiatr Dis Treat 18 (2022) 2813-2835.
[3]U. Petriti, D.C. Dudman, E. Scosyrev, S. Lopez-Leon, Global prevalence of Rett syndrome: systematic review and meta-analysis, Syst Rev 12(1) (2023) 5.
[4]J.L. Neul, P. Fang, J. Barrish, J. Lane, E.B. Caeg, E.O. Smith, H. Zoghbi, A. Percy, D.G. Glaze, Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome, Neurology 70(16) (2008) 1313-21.
[5]J.L. Neul, J.B. Lane, H.S. Lee, S. Geerts, J.O. Barrish, F. Annese, L.M. Baggett, K. Barnes, S.A. Skinner, K.J. Motil, D.G. Glaze, W.E. Kaufmann, A.K. Percy, Developmental delay in Rett syndrome: data from the natural history study, J Neurodev Disord 6(1) (2014) 20.
[6]J.D. Lewis, R.R. Meehan, W.J. Henzel, I. Maurer-Fogy, P. Jeppesen, F. Klein, A. Bird, Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA, Cell 69(6) (1992) 905-14.
[7]P.D. Stenson, E.V. Ball, M. Mort, A.D. Phillips, J.A. Shiel, N.S. Thomas, S. Abeysinghe, M. Krawczak, D.N. Cooper, Human Gene Mutation Database (HGMD): 2003 update, Hum Mutat 21(6) (2003) 577-81.
[8]J. Guy, B. Hendrich, M. Holmes, J.E. Martin, A. Bird, A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome, Nat Genet 27(3) (2001) 322-6.
[9]R.Z. Chen, S. Akbarian, M. Tudor, R. Jaenisch, Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice, Nat Genet 27(3) (2001) 327-31.
[10]L.M. Lombardi, S.A. Baker, H.Y. Zoghbi, MECP2 disorders: from the clinic to mice and back, J Clin Invest 125(8) (2015) 2914-23.
[11]N. Panayotis, Y. Ehinger, M.S. Felix, J.C. Roux, State-of-the-art therapies for Rett syndrome, Dev Med Child Neurol 65(2) (2023) 162-170.