

Potential Liver Threat: Hepatolenticular Degeneration
Introduction: Hepatolenticular Degeneration, commonly known as Wilson's Disease (WD), is a genetic disorder of copper metabolism, inherited in an autosomal recessive manner. Although its prevalence varies among ethnicities and regions, East Asian countries report significantly higher incidence rates than Western nations. A comprehensive study in Anhui Province, China, found the incidence to be approximately 1.96 per 100,000. Fortunately, with timely treatment, this disease is entirely curable, leading to better outcomes.[1]
Pathogenesis:
1. Copper Homeostasis: On average, humans consume about 1.5-5 mg of copper daily. Around 50%-60% of this intake remains unabsorbed and is eliminated through feces, while 25%-40% is absorbed in the duodenum and stored in intestinal cells. Given that copper is vital for many physiological processes, its levels must be strictly regulated.
Copper Molecular Chaperones ensure the delivery of copper to designated intracellular targets. Notable examples include the copper molecular chaperone for SOD, which ferries copper to SOD, and a set of chaperone proteins in the mitochondria, such as Cos11, Cos17, SCO1, and SCO2, responsible for copper delivery to cytochrome oxidase COX. In the Trans-Golgi network, ATOX1 facilitates copper transportation to ATPase proteins: ATP7A and ATP7B.
ATP7B plays a pivotal role in:
l Transporting copper to the plasma's ceruloplasmin within the TGN.
l Releasing excess copper from the cytoplasm, when copper ion levels surge, into vesicles that excrete it into bile.
Throughout this mechanism, ATP7B utilizes the energy from ATP hydrolysis for membrane copper translocation.
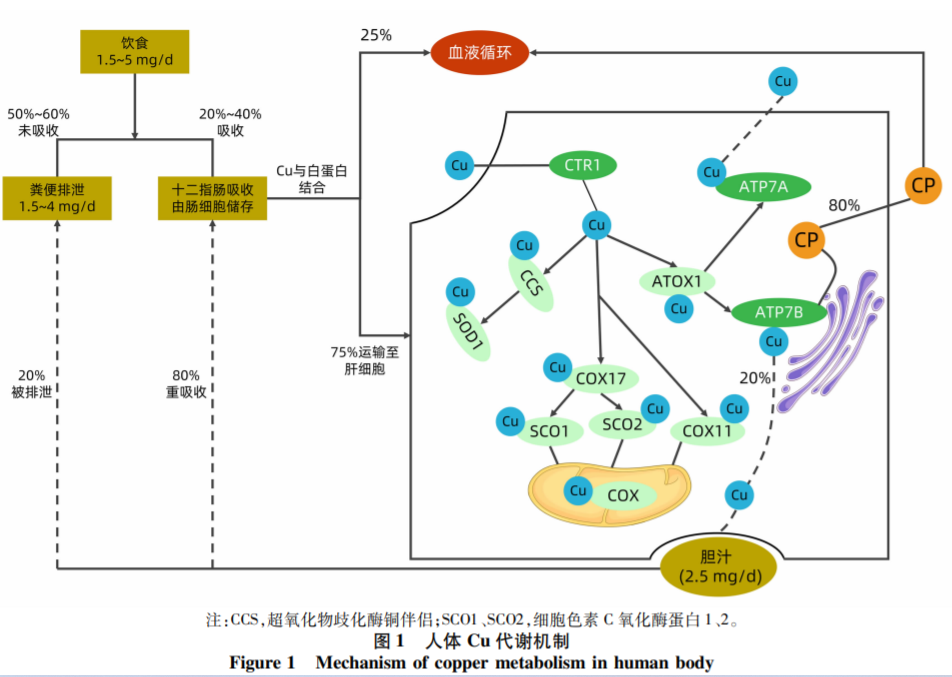
ATP7B gene mutations, seen in WD patients, can reduce the synthesis of ceruloplasmin and hinder biliary copper excretion, resulting in excess copper in tissues like the liver, brain, kidneys, and cornea. This excessive copper leads to liver cell death and copper leakage into the bloodstream, causing damage to various organs and a cascade of clinical symptoms.[2]
Pathogenic Gene:
The ATP7B protein, part of the P-type ATPase superfamily, is encoded by the ATP7B gene on chromosome 13 (13q14.3-q21.1). The gene spans roughly 85 b.
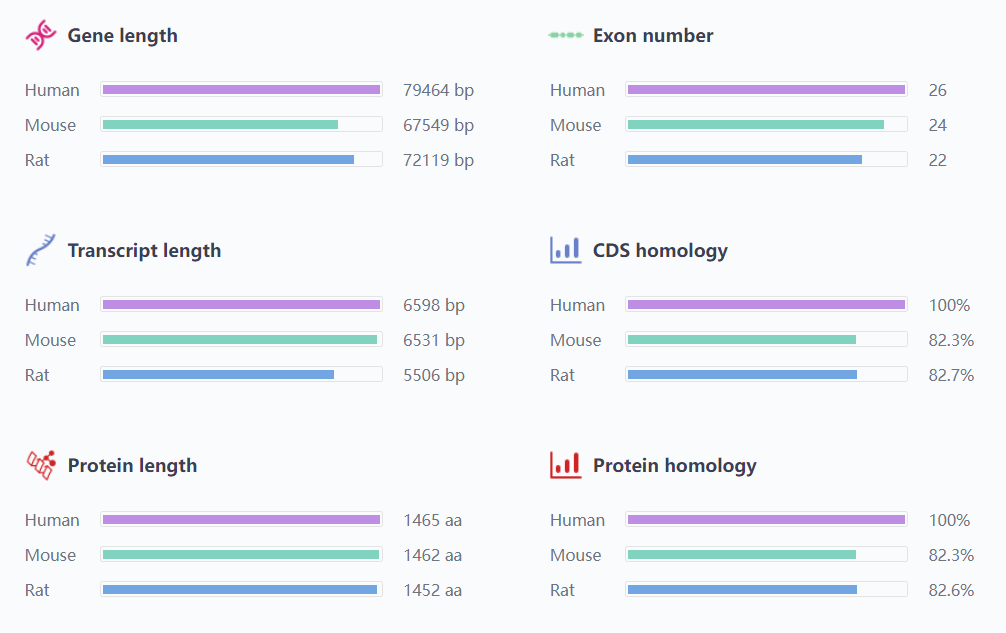
Figure 2. Basic Information of the Pathogenic Gene ATP7B
Wilson's Disease (WD) presents with a range of intricate clinical manifestations. To date, over 1,000 ATP7B mutations have been discovered, of which 380 are confirmed to play a role in the disease mechanism. Predominantly, these mutations are missense types, leading to varied consequences on the ATP7B protein's structure — such as loss of integrity, misfolding, and impaired protein-protein interactions — and its function, impacting processes like phosphorylation, copper ion transport, ATP binding affinity, and intracellular transport.
Two prevalent mutations in European and Asian demographics are p.His1069Gln and p.Arg778Leu. Compound heterozygotes for these mutations are found more frequently than homozygotes. A study spearheaded by Zhang et al.,[16] encompassing 1,366 Chinese WD patients, pinpointed 294 potential pathogenic ATP7B variants. This includes 116 newly identified variants, with 48 classified as "pathogenic variants." The most frequent of these variants were c.2333G > T (p.Arg778Leu), c.2975C > T (p.Pro992Leu), and c.2621C > T (p.Ala874Val).[15]
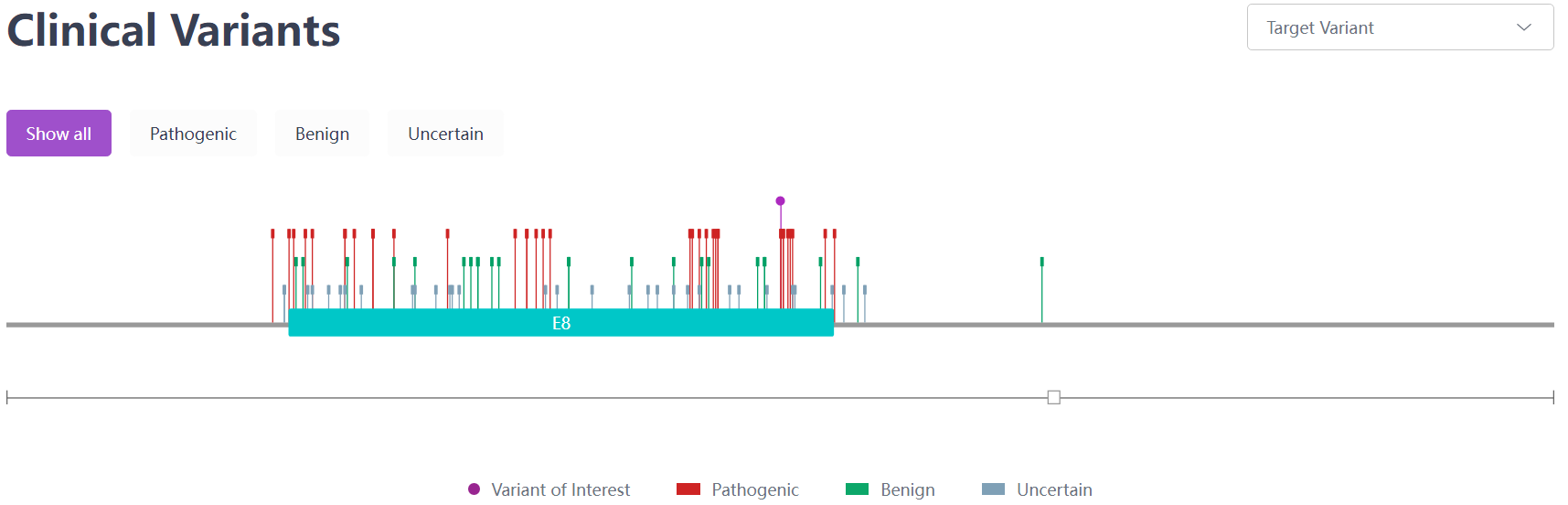
Figure 3. Clinical Mutation c.2333G > T (p.Arg778Leu) sourced from the RDDC database.
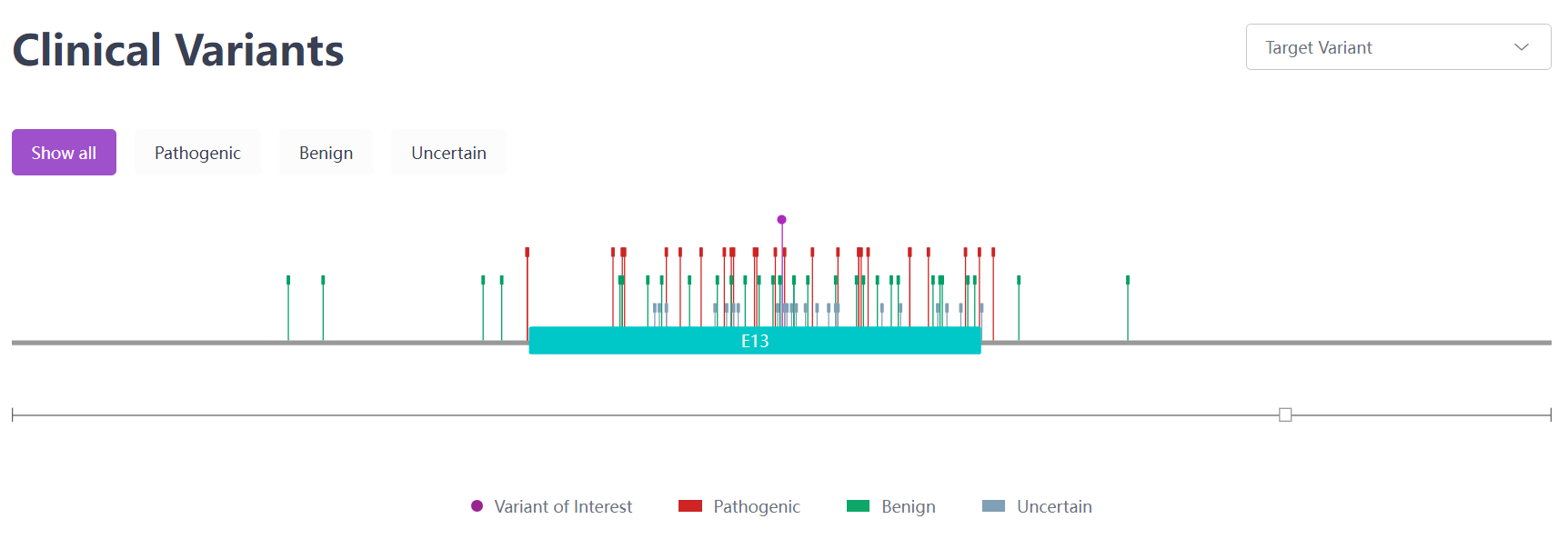
Figure 4. Clinical Mutation c.2975C > T (p.Pro992Leu) sourced from the RDDC database.
Clinical Symptoms:
WD patients display a range of symptoms, mainly affecting the liver and nervous system, but also presenting abnormalities in eyes, kidneys, joints, and other areas.
l Liver: The liver, one of WD's primary targets, exhibits symptoms early in life (>2 years). Manifestations vary from asymptomatic microstructural changes to severe conditions like hepatitis, jaundice, cirrhosis, and acute liver failure.
l Nervous System: Neurological signs, appearing around a decade after liver symptoms, primarily concern the extra-pyramidal motor system. Patients may exhibit muscle tone issues, tremors, stiffness, slow movement, and behavioral anomalies. Some also show chorea-like movements, limb dyskinesia, ataxia, and other uncommon neurological issues.
l Eyes: A classic feature of WD is the K-F ring, a copper deposition in the corneal layer, presenting as a green-brown or dark brown hue. Another eye-related sign is the Kayser-Fleischer ring due to copper accumulation in the lens, though this is rarer.
l Hemolysis: Around 1% of WD patients may exhibit Coombs-negative hemolytic anemia caused by copper-induced damage to red blood cell membranes. In those with jaundice, hemolytic anemia accounts for 28% of cases.
Other Manifestations:
l Kidneys: Damage primarily involves tubular injury, presenting as microscopic hematuria or kidney stones. Glomerular damage is a complication.
l Joints: Rare in WD, joint conditions can mimic other diseases, making them challenging to diagnose. Symptoms can range from osteoporosis and osteomalacia to premature osteoporosis and knee and wrist degenerative joint diseases.
l Cardiac and Endocrine Systems: Rarely, WD might induce myocarditis, arrhythmias, endocrine abnormalities, amenorrhea, miscarriages, male gynecomastia, testicular atrophy, and pancreatitis. Skin manifestations like nail blue nodules and blackberry dermatitis are also infrequent but possible.
Diagnostic Tests
Laboratory Tests:
1) Routine Tests:
l Blood system anomalies might result in anemia, reduced white blood cells, and decreased platelets.
l Liver function anomalies can manifest as elevated liver enzymes, increased bilirubin and bile acids, prolonged clotting time, and hypoproteinemia.
l Renal anomalies might be indicated by hematuria and proteinuria.
2) Copper Metabolism-Related Tests:
l Most patients have blood copper levels below 0.2g/L. A level below 0.1g/L strongly suggests hepatic and basal ganglia degeneration.
l For adults, a 24-hour urine copper level above 100g is a diagnostic criterion; for children, a level above 40g is.
Imaging Tests:
1) B-mode ultrasound:
l Mild cases might only reveal variations in density or uneven liver parenchyma.
l Other findings can include enlarged liver spots, liver enlargement, nodular changes, splenomegaly, and cirrhosis.
2) Head MRI:
l Around 85% of patients with neurological symptoms show head MRI abnormalities, predominantly in the basal ganglia.
l Abnormal signals can also manifest in the midbrain, pons, thalamus, cerebellum, and frontal lobe cortex. Some patients might display widened brain fissures, enlarged ventricles, or MRI changes even before neurological symptoms arise.
Ophthalmologic Slit-Lamp Exam:
l This detects copper deposits on the elastic layer behind the cornea, forming a K-F ring. Studies show K-F ring positivity rates of 55% in patients with hepatic basal ganglia degeneration and up to 90% in those with neurological symptoms. Among asymptomatic patients, this rate is much lower.
Pathological Examination:
l Liver biopsy: Early histological signs in the liver encompass mild fatty degeneration, liver cell glycogenation, and localized liver cell necrosis. As the disease advances, fibrosis and cirrhosis can develop. Patients with hepatic basal ganglia degeneration typically have liver copper content exceeding 250ug/g, but the copper distribution within the liver varies. Therefore, the biopsy site can influence copper content measurements.
Differential Diagnosis
Diagnosing hepatic basal ganglia degeneration is challenging, and it often gets mistaken for other conditions.
Professor Li Xinhua, an expert in this field, highlighted the complexity through the case of Xiao He. Despite being ill for five years and undergoing numerous tests across various hospitals, Xiao He's condition remained undiagnosed. Professor Li, however, managed to diagnose Xiao He with CTLN2, using a holistic approach combining past medical records, recent data, and clinical features. With a new treatment regimen, Xiao He's condition improved remarkably, underscoring the need for a comprehensive diagnostic strategy.
For hepatic basal ganglia degeneration, multiple aspects must be considered:
l Clinical symptoms
l Auxiliary exams
l Genetic analysis
Primary diagnostic indicators include:
l Low levels of serum copper blue protein
l High levels of 24-hour urine copper
l Identification of pathogenic mutations in the ATP7B gene
For patients with predominant liver symptoms, consider differential diagnosis for:
l Chronic viral infections
l Autoimmune hepatitis
l Non-alcoholic liver cirrhosis
l Drug-induced liver damage
l HFE-related primary hemochromatosis
l α1-antitrypsin deficiency
l Alcoholic liver disease, etc.
For patients with primary neurological symptoms, differentiation should be considered for:
l Parkinson's disease
l Menkes syndrome
l Genetic copper blue protein deficiency
l Huntington's disease
l Central nervous system tumors, etc.
Animal Models for Wilson's Disease (WD)
In the continuous endeavor to understand and eventually treat Wilson's Disease (WD), a variety of animal models have emerged as crucial players. These models not only provide a closer look at the progression of the disease but also pave the way for potential therapeutic interventions. Here's a comprehensive breakdown:
1. Mouse Models
1) Toxic Milk (TX) Mice:
l Background: First introduced by Professor Rauch in 1983, TX mice emerged due to a spontaneous mutation within the DL mouse strain. They were named 'toxic milk mice' by Rauch, attributing to the fatal effects of maternal lactation.
l Characteristics: These mice display distinct phenotype features such as diminished pigmentation, developmental anomalies, tremors, and aberrant motor actions. They carry the same gene mutation as seen in WD patients.
l Research Significance: Because they exhibit pronounced liver damage, TX mice serve as invaluable subjects in WD research.
2) Jackson Laboratory Toxic Milk (TX-J) Mice:
l Background: Identified in 1987 within the C3H/HeJ animal resource group at Maine's Jackson Laboratory, TX-J mice are sometimes termed as Jackson's toxic milk mice.
l Genetic Traits: Their genetic anomaly stems from a spontaneous mutation (G712D missense mutation) at position 2135 in exon 8, external to the Atp7B gene. This mutation varies slightly from the one found in TX mice.
l Characteristics: By 3 months, TX-J mice present with an accumulation of copper in the thalamus and basal ganglia. As this deposition persists, elevated copper concentrations are noted in the cerebral cortex.
l Research Significance: The early onset of copper accumulation renders the TX-J mice as exemplary models for scrutinizing copper metabolism and other related subjects.
3) ATP7B Gene Knockout (ATP7B-/-) Mice:
l Development: These are transgenic mice developed using homologous recombination techniques to remove exon 2 from the ATP7B gene. Their use in WD research has been on the rise.
l Characteristics: Predominantly, these mice display reduced ceruloplasmin, augmented copper excretion in urine, and accumulation of copper in the liver. They tend to manifest earlier and more severe liver damage than other models.
l Progression: Starting with early cellular changes and mild inflammation by 1.5 months, they progress to more severe conditions like hepatitis and inflammation by 3-5 months. By 9 months, there are evident signs of bile duct growth, fibrosis, and even tumor formations. Impressively, many of these mice exhibit substantial liver regeneration post severe hepatitis.
l Research Significance: ATP7B-/- mice not only offer insights into the pathology of copper toxicity but also illuminate potential compensatory and defensive strategies against copper toxicity.
2. Rat Models
1) Long-Evans Cinnamon (LEC) Rats:
l Background: Derived from the Long-Evans rat population, LEC rats are inbred strains bearing mutations. These rats remarkably have a genetic homology with humans exceeding 80%.
l Characteristics: They primarily exhibit symptoms akin to Wilson's disease, such as spontaneous liver copper accumulation, reduced serum copper, and ceruloplasmin (blue proteinemia).
l Clinical Manifestations: Around the age of 4 months, these rats can suffer acute liver failure or hepatitis, marked by raised levels of transaminases and bilirubin. Additionally, they face partial to extensive hepatocyte necrosis, leading to mortality rates hovering between 30%-40%.
l Progression: Those that survive past this acute phase might later, around 12 months, manifest conditions like liver fibrosis, hepatocellular carcinoma, and cholangiocarcinoma.
l Research Value: Due to their propensity for swift liver disease progression and high mortality rates by 4 months of age, LEC rats serve as ideal models for intervention studies in Wilson's disease.
2) PINA and ATP7B gene-deficient (LPP) Rats:
l Development: To discern and isolate the impacts of ATP7B mutations, Ahmed and colleagues interbred LEC rats with PVG rats. These PVG rats, being wild-type, carry the NAT (N-acetyltransferase) and ATP7B genes and have the usual fur color. The result of this breeding was the LPP rat strain, characterized by a regular NAT and fur color but with an ATP7B gene mutation.
l Characteristics: Right from birth, LPP rats start accumulating copper in their livers. By the 90-day mark, they begin to show liver biochemical anomalies.
l Clinical Manifestations: If the liver disease escalates quickly and remains untreated, these LPP rats may face mortality within just 30 days.
3. Large Mammals
1) Bedlington Terrier:
l Symptoms: Bedlington Terriers are known to accumulate copper in the liver, leading to damage characterized by conditions such as fibrosis and cirrhosis. This mirrors some symptoms observed in Wilson's disease patients.
l Usage: These terriers have been pivotal in copper loading studies. [7]
l Genetic Factors: Interestingly, the genetic anomaly in these canines is attributed to the COMMD1 gene, differing from the ATP7B gene commonly linked with Wilson's disease. [8]
l Limitations: Although the liver symptoms align with WD, Bedlington Terriers neither display neurological defects as some rodent models do nor show abnormal levels of ceruloplasmin. Given these differences, their application in WD research is less frequent.
2) Labrador Retriever:
l Research Findings: A 2016 genome-wide association study (GWAS) shed light on Labrador Retrievers, revealing a mutation from arginine to glutamine in the ATP7B gene's C-terminal. This mutation correlated with heightened liver copper levels.[9]
l Significance: Due to these findings, Labrador Retrievers have been introduced as an additional animal model for investigating Wilson's disease.
3) North Ronaldsay Sheep:
l Attributes: These sheep are intriguing for their observed copper accumulation in both the liver and the nervous system.
l Comparison: In terms of clinical and genetic presentation, North Ronaldsay sheep differ from typical WD manifestations, akin to the canine models. [10]
l Genetics: The precise genetic anomaly in this sheep model is yet to be discerned.
l Research Value: Despite certain distinctions, these sheep are invaluable, particularly when exploring copper accumulation in the brain. [11]
Treatment Guidelines for Wilson's Disease
The cornerstone of Wilson's disease treatment is promoting copper excretion and reducing copper absorption, aiming for a negative copper metabolic balance. Treatment strategies encompass dietary measures, medications, and in severe cases, liver transplantation. Early initiation of treatment results in minimized damage and improved prognosis.
1. Dietary Approach:
l Low Copper Diet: It's crucial to restrict the intake of high-copper foods. This primarily includes avoiding:
n Nuts
n Peas
n Shellfish
n Chocolate
2. Pharmacological Interventions:
l D-Penicillamine: Introduced in 1956, this remains a primary treatment, particularly favored in developing countries. It's suitable for diverse clinical manifestations of WD.
l DMPS (Sodium 2,3-dimercaptopropane-1-sulfonate): This chelating agent, known for its two sulfhydryl groups and good water solubility, facilitates the excretion of heavy metals. Initially employed in China for Wilson's disease (WD) treatment, DMPS boasts a copper-chelating effect that's 2.6 times stronger than D-Penicillamine. It also exhibits fewer side effects, such as aggravation of neurological symptoms. It's especially beneficial for severe WD cases — including those with acute liver failure, neurological challenges, or those who don't respond well to D-Penicillamine. DMPS can be used alongside or in rotation with zinc agents and D-Penicillamine.
l DMSA (Succimer): This broad-spectrum chelator, developed by the Shanghai Institute of Materia Medica, has a milder copper-chelating capacity compared to D-Penicillamine. However, its ability to cross the blood-brain barrier due to its lipophilicity aids in alleviating neurological and psychiatric symptoms in WD patients. It's recommended for patients with diverse liver damages, neurological issues, or those allergic to D-Penicillamine. It can also be combined with or rotated between zinc agents and D-Penicillamine.
l Trientine Tetrahydrochloride (Cuvrior™): This oral trientine variant is recognized in the U.S. as an orphan drug for treating WD patients intolerant to D-Penicillamine. Recently, the China National Medical Products Administration endorsed its use, signaling its potential market launch in China by 2023.
l Tetrathiomolybdate (TM/Decuprate): Developed from tetramolybdate and choline, TM is an upcoming copper reduction therapy for Wilson's disease, currently being pursued by Wilson Therapeutics AB under the codename WTX101. Recognized as an orphan drug in both the U.S. and the European Union, it shows promise as a potent WD treatment.
l Zinc Agents: Compounds such as zinc sulfate, gluconate, and acetate are employed to mitigate free copper levels in the bloodstream, thus playing a pivotal role in Wilson's disease therapy.
l Dimercaprol (BAL): Also termed British anti-Lewisite, this FDA-approved chelator is occasionally essential for detoxifying a subset of WD patients.
For varied neurological and psychiatric symptoms, treatments can be supplemented with medications such as trihexyphenidyl, levodopa, benzodiazepines, or antipsychotics.
References:
[1] https://lczl.med.wanfangdata.com.cn/Home/DiseaseDetail?Id=JB23049
[2] 黄稚真,胡敏,梁惠卿,艾兆秀 & 孙雨洁.(2023).化痰祛瘀法治疗肝豆状核变性研究进展. 陕西中医(06),815-817.
[3] RAUCH H. Toxic milk, a new mutation affecting cooper metabolism in the mouse[J]. J Hered, 1983, 74(3): 141-144.
[4] PRZYBYŁKOWSKI A, GROMADZKA G, WAWER A, et al. Neurochemical and behavioral characteristics of toxic milk mice: an animal model of Wilson's disease[J]. Neurochem Res, 2013, 38(10): 2037-2045.
[5] AHMED S, DENG J, BORJIGIN J. A new strain of rat for functional analysis of PINA[J]. Brain Res Mol Brain Res, 2005, 137(1-2): 63-69.
[6] ZISCHKA H, LICHTMANNEGGER J, SCHMITT S, et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease[J]. J Clin Invest, 2011, 121(4): 1508-1518.
[7] FIETEN H, PENNING LC, LEEGWATER PA, et al. New canine models of copper toxicosis: diagnosis, treatment, and genetics[J]. Ann N Y Acad Sci, 2014, 1314: 42-48.
[8] HAYWOOD S, VAILLANT C. Overexpression of copper transporter CTR1 in the brain barrier of North Ronaldsay sheep: Implications for the study of neurodegenerative disease[J]. J Comp Pathol, 2014, 150(2-3): 216-224.
[9] FIETEN H, GILL Y, MARTIN AJ, et al. The Menkes and Wilson disease genes counteract in copper toxicosis in Labrador retrievers: A new canine model for copper-metabolism disorders[J]. Dis Model Mech, 2016, 9(1): 25-38.
[10] HAYWOOD S, MVLLER T, MACKENZIE AM, et al. Copper-induced hepatotoxicosis with hepatic stellate cell activation and severe fibrosis in North Ronaldsay lambs: A model for non- Wilsonian hepatic copper toxicosis of infants[J]. J Comp Pathol, 2004, 130(4): 266-277.
[11] HAYWOOD S, VAILLANT C. Overexpression of copper transporter CTR1 in the brain barrier of North Ronaldsay sheep: Implications for the study of neurodegenerative disease[J]. J Comp Pathol, 2014, 150(2-3): 216-224.
[12] 肝豆状核变性诊疗指南(2022版)
[13] https://mp.weixin.qq.com/s/Osr1e_I5W10WrUp1swHX1A
[14] http://www.100pei.com/5029.html
[15] 玛力帕提·艾尔肯江,凯迪日亚·库尔班,徐玲,孙晓风.肝豆状核变性发病机制、临床表型-基因型关系及药物治疗研究进展[J].临床肝胆病杂志,2023,39(6):1497-1504.
[16] ZHANG S,YANG W,LI X,et al. Clinical and geneticcharacterization of a large cohort of patients with Wilson ' sdisease in China[ J]. Transl Neurodegener, 2022,11( 1) : 13.