

肝豆状核变性——肝脏中的潜在威胁
肝豆状核变性(Hepatolenticulardegeneration,HLD),又名威尔逊病、Wilson病(Wilson disease,WD),是一种遗传性铜代谢异常的肝脏疾病,呈常染色体隐性遗传。本病的发病率存在人种和地域的差异,东亚国家明显高于欧美国家。我国安徽省的一项大样本人群WD流行病学调查显示该病发病率约为1.96/10万。本病是完全可以治愈的,治疗开始愈早,预后愈好[1]。
致病机理
铜的稳态
人们每天从饮食中摄入1.5~5mg的铜元素(Cu),其中 50% ~ 60%未吸收的 Cu 随粪便排出;25% ~40% 从十二指肠吸收,由肠细胞储存。Cu是几乎所有生物体的基本元素,Cu是多种关键代谢酶的辅助因子,这些酶可驱动广泛的生理过程。因此,全身铜水平变化必须维持在一个很小的范围内,以确保正常的生化过程。
Cu的分子伴侣将Cu运送到特定的细胞内靶点,例如:SOD的Cu分子伴侣将Cu运送到SOD;线粒体中的一组分子伴侣蛋白( Cos11、Cos17、SCO1 和SCO2),将Cu传递给细胞色素氧化酶COX;在反式高尔基网络(Trans-Golgi network,TGN)中,Cu转运蛋白(ATOX1)将Cu转运至Cu转运 ATP酶1(ATP7A)和跨膜 Cu 转运 ATP 酶2(ATP7B)。
ATP7B 有助于将 Cu 转运到 TGN 和 Cu 的胆道排泄。在肝细胞内,ATP7B 在 TGN 或细胞质囊泡中发挥两种重要功能。 在 TGN 中,ATP7B 通过将6个Cu分子运输到原血浆CP中,激活功能性 CP。在细胞质中,当细胞内 Cu 离子水平升高时,ATP7B 将多余的 Cu 释放到囊泡中,并通过胞外排出,穿过根尖管膜进入胆汁。在这一过程中,ATP7B 借助 ATP 水解释放的能量磷酸化,随后脱磷酸化释放 Cu 跨膜所需的能量。
因此,ATP7B依赖性胆汁 Cu 排泄是维持 Cu 代谢平衡的主要方式。
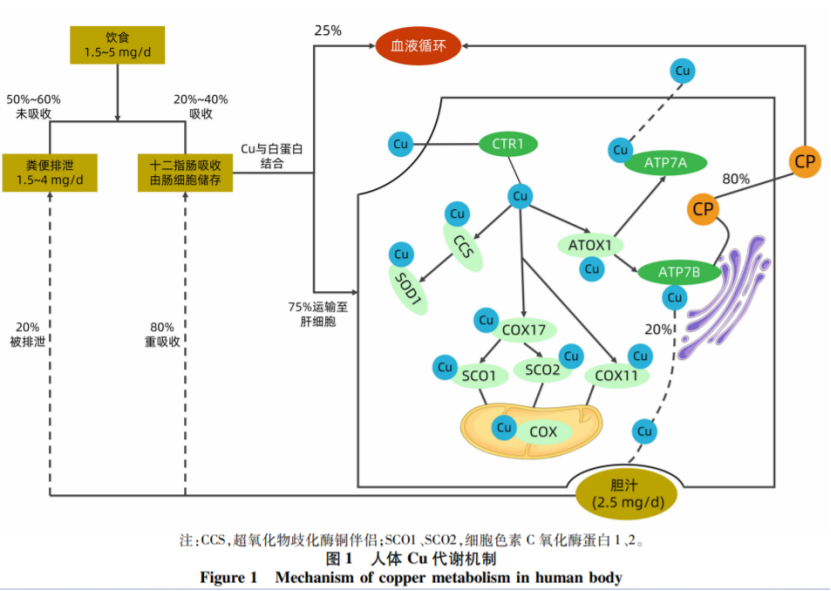
WD患者ATP7B基因变异导致 ATP7B 酶活性改变,由此导致致使铜蓝蛋白合成量减少、胆道排铜出现障碍,过量的铜蓄积在肝、脑、肾、角膜等组织,造成肝细胞死亡和铜渗漏入血,进一步引起肝、脑、眼睛、肾脏等组织器官的损伤,引起一系列临床症状[2]。
致病基因
ATP7B 蛋白属P型ATP 酶超家族,由位于第13号染色体长臂(13q14.3 ~q21.1),全长约 85 b 的 ATP7B 基因编码。

图2 致病基因ATP7B基本信息 来源:RDDC数据库
WD具有复杂的临床表现,目前已发现超过1000种ATP7B突变。其中380个已经证实与疾病发病机制有关。大多数是错义突变,这些突变对 ATP7B 蛋白结构(例如,ATP7B 完整性的丧失、错误折叠、蛋白间相互作用受损)和功能(磷酸化、Cu离子运输异常、ATP 结合亲和力下降和细胞内运输异常)产生了不同程度的影响。
在欧洲和亚洲人群中,最常见的突变分别是p.His1069GIn和 p.Arg778Leu,其中复合杂合子比纯合子更为常见。在Zhang 等[16]发表的一项共纳入1366例中国WD 患者的评估基因型-表型相关性的研究鉴定出了294 例潜在致病性 ATP7B 变异,包括 116 例新变异,其中48 个可归为“致病变异”。变异频率依次为c.2333G >T (p.Arg778Leu)、c.2975C >T (p. Pro992Leu)、c.2621C >T (p.Ala874Val)[15]。
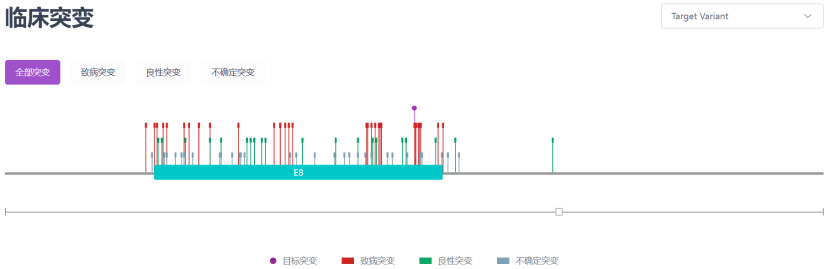
图3:c.2333G >T (p.Arg778Leu)临床突变 来源:RDDC数据库
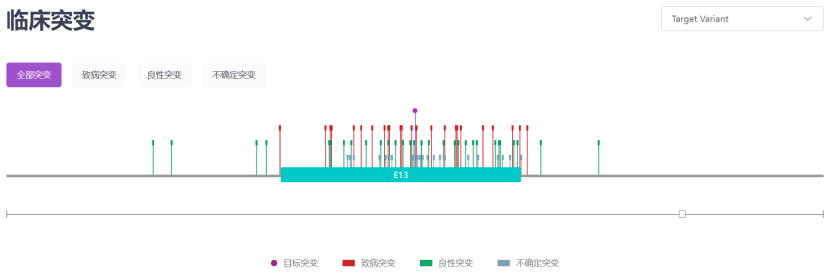
图4:c.2975C >T (p. Pro992Leu) 临床突变 来源:RDDC数据库
临床症状
WD患者临床表现多样,因受累器官和程度不同而异主要表现为肝受累和神经系统受累。此外,还可出现眼部异常、溶血、肾脏损伤、骨关节异常等多种临床表现。
肝脏表现:肝脏是 WD 最常累及的器官之一,发病相对较早 (>2 岁就可能发病),肝受累在临床上表现为一系列广泛的症状和体征,包括无症状的肝脏细微形态学改变、急性单纯性自限性肝炎、重型肝炎、复发性黄疸(存在溶血)、肝硬化伴或不伴门脉高压症,甚至急性肝衰竭(ALF)。
神经系统表现:神经系统病变常较肝病晚 10 年出现(通常 > 15 岁) ,WD 神经系统表现多种多样,但大多为锥体外系功能障碍,常见表现有:肌张力障碍、震颤、肢体僵硬和运动迟缓、精神行为异常,少数患者可出现舞蹈样动作、手足徐动、共济失调等其他少见的神经症状。
眼部表现:K-F 环是铜沉着于角膜后弹力层而形成的绿褐色或暗棕色环,是 WD 的典型特征之一。葵花样白内障是 WD 的另一个眼部表现,为铜沉积于晶状体所致,较为少见。
溶血:WD 可因过多的铜离子损伤红细胞膜而发生 Coombs 阴性的溶血性贫血。溶血性贫血可急性发作,也可呈阵发性或慢性病程。小样本研究结果显示,约 1% 的 WD 患者以溶血性贫血为首发表现,有黄疽的 WD 患者中,溶血性贫血占28%。
其他表现:WD 还可以引起肾脏、骨关节等其他器官组织损害。
肾损伤以肾小管损伤为主,可表现为镜下血尿和肾结石,为近曲小管和远曲小管上皮中的铜沉积损伤肾小管上皮细胞所致;肾小球损伤更多见于整合剂治疗的并发症。
骨关节病并不常见,可表现为骨质疏松症、骨软化症、自发性骨折、佝偻病、剥脱性骨软骨炎、骸骨软骨软化症、过早骨质减少以及膝盖和手腕的退行性关节炎等,与其他骨关节病在临床表现上难以区分。
此外,还可以引起心肌炎、心律失常等心脏损伤,女性闭经、流产,男性乳房发育、睾丸萎缩、甲状旁腺功能减退、胰腺炎等内分泌异常,偶见指甲蓝色隆突和黑棘皮病等。
一些学者还探究了突变基因与疾病表型的关系,如图5:

辅助检查
1.实验室检查
(1)常检
若累及血液系统,可出现贫血、白细胞下降、血小板下降;肝功能检查可见肝酶升高、胆红素升高、胆汁酸升高、凝血时间延长和低蛋白血症等,肾脏检查可见血尿、蛋白尿等。
(2)铜代谢相关检查
绝大多数患者血铜蓝蛋白<0.2g/L,如果<0.1g/L强烈提示肝豆状核变性。24小时尿铜在成人患者中>100g为诊断标准之一,在儿童患者中>40g为诊断标准之一。
2.影像学检查
(1)B超
轻者仅表现为密度增强、减低或不均,还可以表现为肝实质光点增粗、肝脏增大,甚至结节状改变、脾大等肝硬化表现。
(2) 头核磁约85%神经型患者头颅核磁显示异常,主要累及基底节,也可出现中脑和脑桥、丘脑、小脑及额叶皮质等部位的异常信号,还可有不同程度的脑沟增宽、脑室扩大等。在神经系统症状出现之前,部分患者也可出现头颅核磁的异常改变。
3.眼科裂隙灯检查
由于铜沉积于角膜后弹力层,在角膜与巩膜的内表面上出现绿色或金褐色的角膜色素环,即K-F环。有研究提示,肝脏型肝豆状核变性患者中K-F环阳性率为55%,在神经型患者中K-F环阳性率高达90%。在儿童症状前患者中,K-F环阳性率较低。
4.病理学检查
肝脏活检:肝脏最早的组织学异常包括轻度脂肪变性、肝细胞内糖原化和局灶性肝细胞坏死。伴随着病程进展,可出现纤维化、肝硬化。肝豆状核变性患者肝铜含量>250ug/g (千重),但铜在肝脏中分布不均,铜含量测定可能会受所取标本的影响。
诊断鉴别
肝豆状核变性的诊断较为复杂,常常被误诊为其他疾病。
在实际工作中,肝豆状核变性的专家李新华教授就曾接手过一例发病5年、辗转多家医院、做过许多检查仍无法确诊的患者(化名小何)。李教授分析了病人的病例特点,结合外院及急诊科资料,临床诊断小何得的是CTLN2。据此,采取了不一样的治疗:停止含糖补液、补充脂肪乳及蛋白质、精氨酸静滴等治疗。入院当天晚上患者的神志就恢复了正常,可以自主交流。随后小何的血尿质谱与基因检测结果出来,佐证了李教授的诊断。
因此,对于肝豆状核变性的诊断鉴别应多方面综合分析,结合临床表现、辅助检查及基因分析来下论断。肝豆患者铜蓝蛋白降低、24小时尿铜增高和ATP7B 基因检出致病突变有利于和其他疾病鉴别。
对于肝脏受累为主的患者,应与慢性病毒性感染、自身免疫性肝炎、非精性肝硬化、药物性肝损、原发性硬化性胆管炎、HFE相关的原发性血色素沉着症、a1抗胰蛋白缺乏症和酒精性肝病等鉴别。
对于神经系统受累为主的患者,应与帕金森病、Menkes综合征、遗传性铜蓝蛋白缺乏症、MEDNIK综合征、肌张力障碍、亨廷顿病、原发性震颤、神经退行性病变、中枢神经系统肿瘤及其他遗传代谢病鉴别。
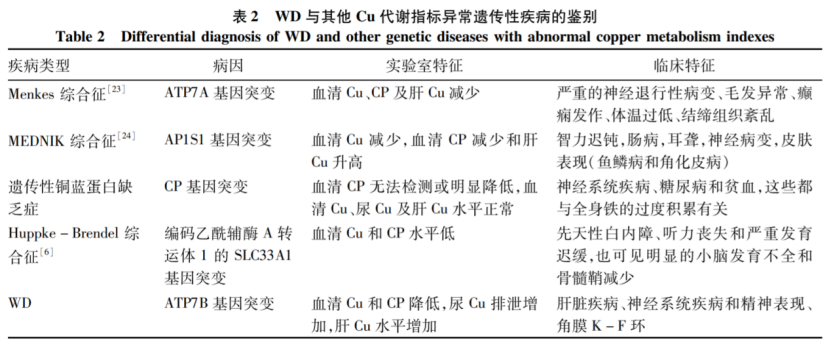
图6:WD与其他Cu代谢指标异常遗传性疾病的鉴别 来源:肝豆状核变性发病机制、临床表型-基因型关系及药物治疗研究进展
动物模型
(1) 小鼠
毒乳(Toxic milk,TX)小鼠:TX小鼠是由DL品系小鼠自然突变产生,由Rauch教授[3]于1983年首次提出。其表型特征为: 色素沉着减少、生长发育不良、震颤、行为运动异常。由于母鼠母乳喂养的致命影响,Rauch将这种新的突变品系命名为毒乳小鼠。由于TX小鼠与WD患者突变基因相同且肝损伤表现突出,目前已广泛应用于WD研究。
Jackson毒乳(the Jackson laboratory toxic milk,TX-J)小鼠:TX-J小鼠,又被称为Jackson毒乳小鼠,是在1987年缅因州巴尔港杰克逊实验室的C3H/HeJ动物资源群体中发现的一种新的常染色体隐性突变体,其遗传缺陷源于Atp7B基因外显子8中2135位的一个自发隐性点突变,导致G712D错义,这与TX小鼠稍有差异。相关研究发现TX-J小鼠在3个月大的时候就表现出丘脑和豆状核的铜过载。随着铜的不断沉积,大脑皮层中的铜浓度也有所升高[4]。TX-J小鼠是一种较理想的WD模型,由于铜沉积出现较早,因此较适合进行铜代谢等方面研究。
ATP7B基因敲除(ATP7B-/-)小鼠:利用同源重组技术敲除ATP7B基因第2外显子的转基因小鼠也越来越多的被用于WD研究。ATP7B-/-小鼠主要表现有低铜蓝蛋白血症、尿铜增加及肝脏铜沉积,与其他模型相比发病较早且肝损伤更严重。ATP7B-/-小鼠的肝脏病理损伤程度取决于铜暴露的时间和细胞内铜的分布。肝损伤包括早期超微结构改变、脂肪变性和轻度炎症(1.5月龄时),随后出现肝炎、发育不良和晚期坏死性炎症(3~5月龄时)。这些变化伴随的是胆管增生、纤维化的发展,甚至是后期(9月龄)的肿瘤增生。然而,大多数小鼠在严重肝炎中存活下来,并在肝脏的大部分区域表现出显著的再生。因此,ATP7B-/-小鼠不仅是研究铜中毒病理的有效模型,而且为研究铜中毒的代偿机制和防御机制提供了途径。
(2) 大鼠
Long-Evans cinnamon(LEC)大鼠:LEC大鼠是从Long-Evans大鼠群体中分离出来的突变大鼠的自交系, 其与人类基因同源性达80%以上。LEC大鼠主要表现出自发性肝铜异常积聚伴随低血清铜和低铜蓝蛋白,由于此与WD的表现类似,因而被应用于WD的研究。LEC大鼠约4月龄时出现暴发性肝炎或暴发性肝衰竭、转氨酶和胆红素升高及部分或大量肝细胞坏死,病死率>30%~40%,存活下来的大鼠多在12月龄时发生纤维化并伴有肝细胞癌和胆管癌。LEC大鼠是一种铜快速积累模型,在4月龄时由于严重肝炎和之后的肝细胞癌导致病死率很高。由于其肝病进展迅速,因此特别适合用于进行干预研究。
PINA和ATP7B基因有缺陷(LPP)大鼠:为了消除非ATP7B突变对表型的影响,Ahmed等[5]将LEC大鼠与PVG大鼠杂交,后者为野生型,具有NAT(N-乙酰基转移酶)、ATP7B基因和正常毛色,这一过程产生了具有野生型大鼠NAT和毛色但ATP7B发生突变的新鼠种,称为LPP大鼠。LPP大鼠自出生后肝铜逐渐积累,并在90天左右出现肝生化异常,一旦肝疾病迅速进展,未经治疗的LPP大鼠可在30天内死亡[6]。
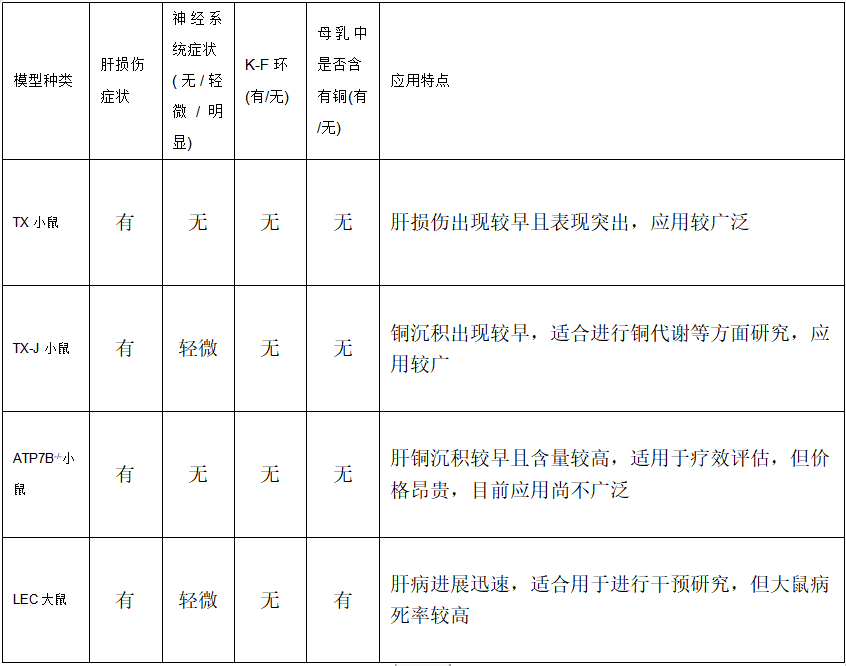
图7:常见WD动物模型特点 来源:来源于肝豆状核变性发病机制、临床表型-基因型关系及药物治疗研究进展
(3) 大型哺乳动物
Bedlington犬:Bedlington犬最早也曾被当作Wilson病的动物模型。由于肝铜的聚集引起肝损伤和继发性溶血并表现出与Wilson病患者相似的肝纤维化和肝硬化的肝铜中毒迹象而曾被用作Wilson病动物模型进行铜负荷的相关研究[7]。然而,其潜在的遗传缺陷是在COMMD1基因中发现的而非ATP7B基因[8]。虽然其肝脏铜的积聚和肝脏病理与WD相似,但与啮齿动物模型一样,不存在明显的神经系统缺陷且铜蓝蛋白浓度正常,故目前该模型较少被使用。
拉布拉多寻回犬:2016年,一项关于拉布拉多寻回犬铜中毒的全基因组关联(GWAS)研究[9]表明,C端ATP7B的精氨酸至谷氨酰胺突变与肝脏铜含量增加有关联,从而将其确立为进一步的Wilson病动物模型。
北罗纳德赛羊:北罗纳德赛羊是另一个有趣的哺乳动物肝脏和神经系统铜积累模型。然而,与犬类模型一样,其在临床上和遗传学上与WD不同[10]。其潜在的基因缺陷目前仍然未知。然而,该模型对于研究铜在大脑中的积累等方面有一定的重要性[11]。
治疗指南
原则是促进铜的排泄及减少铜的吸收,建立铜代谢的负平衡。治疗方式主要是低铜饮食和药物治疗。对于本病所致的急性肝功能衰竭或失代偿性肝硬化患儿,经上述各种治疗无效时可考虑进行肝移植。治疗越早,损害越轻,预后越好[12]。
低铜饮食
避免进食含铜高的食物,如坚果、豌豆、软体动物、巧克力等。
药物盘点
1. D-青霉胺
D-青霉胺是世界上第一个口服的治疗 WD 药物,从 1956 年开始临床应用,目前仍是治疗本病的一线药,适用于各种临床类型的 WD 患者,在发展中国家则为首选药。
2.二疏磺酸钠(DMPS)
DMPS 是含有 2 个疏基的重金属整合剂,水溶性好可显著促进重金属的排泄。我国首先用于治疗 WD,其驱铜作用是 D-青霉胺的 2.6 倍,治疗后神经症状加重等不良反应少于D-青霉胺。适用于 ALF 等重症 WD 患者、神经精神症状的 WD 患者,以及对 D-青霉胺过敏,或 D-青霉胺疗效欠佳需要快速驱铜的患者。可与锌剂联合使用,也可与 D-青霉胺、锌剂交替使用。
3.二疏丁二酸(DMSA)
DMSA 是中国科学院上海研究所研制的广谱重金属整合剂,DMSA 驱铜作用较 D-青霉胺弱,但具有脂溶性,能进入血脑屏障,有助于改善神经精神症状,不良反应相对较少。可用于有不同程度肝损伤,或神经精神症状的WD 患者,以及对 D-青霉胺过敏或不耐受者。可与锌剂联合使用,或与 D-青霉胺、锌剂交替使用。
4. 曲恩汀四盐酸盐Cuvrior™(trientine tetrahydrochloride)
是一种口服曲恩汀制剂,曲恩汀四盐酸盐在美国已被授予孤儿药资格,用于治疗对青霉胺不耐受的威尔逊病患者。2022年9月21日,中国国家药监局药品审评中心(CDE)发布消息,Orphalan公司研发的新型药物四盐酸曲恩汀薄膜衣片在中国获得了优先审批,2023有望获批在中国上市。
5. 四硫钼酸盐(Tetl.athiomolybdate,TM)
双胆碱四硫代钼酸盐(Tiomolibdate,商品名Decuprate),是目前正在研究的四硫钼酸盐和胆碱合成的一种盐,是针对Wilson病的临床开发中的去铜疗法,其代号为WTX101,由Wilson Therapeutics AB提供。Wilson Therapeutics由HealthCap于2012 年创立。Decuprate在美国和欧盟已获得孤儿药地位,作为Wilson病潜在治疗方案。
6. 锌制剂:
硫酸锌、葡萄糖酸锌、醋酸锌等锌盐类药物可降低血液循环中游离铜的水平,促进脑部受累的改善,作为治疗Wilson病方案的一部分[13]。
7. 二巯基丙醇(Dimercaprol)
二巯基丙醇也被称为路易氏剂(BAL),是一种铜螯合剂,已被FDA批准用于治疗Wilson病。少部分患者偶尔需要注射二巯基丙醇解毒[14]。
此外,根据神经精神方面的不同症状可选用苯海索、左旋多巴、安定类或抗精神病药物等。
参考资料:
[1]https://lczl.med.wanfangdata.com.cn/Home/DiseaseDetail?Id=JB23049
[2]黄稚真,胡敏,梁惠卿,艾兆秀 & 孙雨洁.(2023).化痰祛瘀法治疗肝豆状核变性研究进展. 陕西中医(06),815-817.
[3]RAUCH H. Toxic milk, a new mutation affecting cooper metabolism in the mouse[J]. J Hered, 1983, 74(3): 141-144. DOI: 10.1093/oxfordjournals.jhered.a109751.
[4]PRZYBYŁKOWSKI A, GROMADZKA G, WAWER A, et al. Neurochemical and behavioral characteristics of toxic milk mice: an animal model of Wilson's disease[J]. Neurochem Res, 2013, 38(10): 2037-2045. DOI: 10.1007/s11064-013-1111-3.
[5]AHMED S, DENG J, BORJIGIN J. A new strain of rat for functional analysis of PINA[J]. Brain Res Mol Brain Res, 2005, 137(1-2): 63-69. DOI: 10.1016/j.molbrainres.2005.02.025.
[6]ZISCHKA H, LICHTMANNEGGER J, SCHMITT S, et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease[J]. J Clin Invest, 2011, 121(4): 1508-1518. DOI: 10.1172/JCI45401.
[7]FIETEN H, PENNING LC, LEEGWATER PA, et al. New canine models of copper toxicosis: diagnosis, treatment, and genetics[J]. Ann N Y Acad Sci, 2014, 1314: 42-48. DOI: 10.1111/nyas.12442.
[8]HAYWOOD S, VAILLANT C. Overexpression of copper transporter CTR1 in the brain barrier of North Ronaldsay sheep: Implications for the study of neurodegenerative disease[J]. J Comp Pathol, 2014, 150(2-3): 216-224. DOI: 10.1016/j.jcpa.2013.09.002.
[9]FIETEN H, GILL Y, MARTIN AJ, et al. The Menkes and Wilson disease genes counteract in copper toxicosis in Labrador retrievers: A new canine model for copper-metabolism disorders[J]. Dis Model Mech, 2016, 9(1): 25-38. DOI: 10.1242/dmm.020263.
[10]HAYWOOD S, MVLLER T, MACKENZIE AM, et al. Copper-induced hepatotoxicosis with hepatic stellate cell activation and severe fibrosis in North Ronaldsay lambs: A model for non- Wilsonian hepatic copper toxicosis of infants[J]. J Comp Pathol, 2004, 130(4): 266-277. DOI: 10.1016/j.jcpa.2003.11.005.
[11]HAYWOOD S, VAILLANT C. Overexpression of copper transporter CTR1 in the brain barrier of North Ronaldsay sheep: Implications for the study of neurodegenerative disease[J]. J Comp Pathol, 2014, 150(2-3): 216-224. DOI: 10.1016/j.jcpa.2013.09.002.
[12]肝豆状核变性诊疗指南(2022版)
[13]https://mp.weixin.qq.com/s/Osr1e_I5W10WrUp1swHX1A
[14]http://www.100pei.com/5029.html
[15]玛力帕提·艾尔肯江,凯迪日亚·库尔班,徐玲,孙晓风.肝豆状核变性发病机制、临床表型-基因型关系及药物治疗研究进展[J].临床肝胆病杂志,2023,39(6):1497-1504.DOI:10.3969/j.issn.1001-5256.2023.06.037.
[16]ZHANG S,YANG W,LI X,et al. Clinical and geneticcharacterization of a large cohort of patients with Wilson ' sdisease in China[ J]. Transl Neurodegener, 2022,11( 1) : 13. DOl: 10.1186/s40035 -022 -00287 -0.