

Mechanisms of Hereditary Spinocerebellar Ataxia (SCA) [2] - Research
Mechanisms of Hereditary Spinocerebellar Ataxia (SCA) [2] - Research
Disease diagnosis
Before diagnosing a patient with autosomal dominant spino-cerebellar ataxia (SCA) with progressive ataxia, it is necessary to exclude acquired causes through detailed medical history, physical examination, laboratory tests, etc. Given the large overlap and heterogeneity of clinical symptoms in SCA, the diagnosis mainly relies on genetic testing. The diagnostic criteria for SCA are as follows: 1) progressive ataxia is the main clinical manifestation, and acquired causes of ataxia have been excluded; 2) there is a family history of similar diseases in the previous generation; and 3) SCA genetic testing is positive [1].
If the clinical evidence for diagnosing SCA is clear, molecular genetic testing should be initiated. Only in special cases, such as when a specific SCA genotype is known in the family, the clinical phenotype highly indicates a certain SCA (such as vision loss in SCA7), or when a certain SCA has a higher prevalence in a particular region (such as SCA2 in Cuba), targeted single-gene testing is recommended. In other cases, it is recommended to use systematic SCA genetic testing. First, dynamic CAG repeat testing should be performed for polyglutamine SCAs (i.e., SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, DRPLA), as the incidence of these types of SCAs is relatively high. When CAG dynamic repeat testing is negative, appropriate sequencing options can be selected based on the actual situation: 1) Whole-exome sequencing (WES) can detect traditional mutation types of SCA and can also perform reanalysis when new ataxia genes are discovered; 2) As WES cannot detect repeat mutations, dynamic mutation testing for repeated non-coding mutations (i.e., SCA8, SCA10, SCA12, SCA31, SCA36, and SCA37) must be performed to evaluate all genetic causes of SCA; 3) If WES does not include a search for specific deletions, specific testing for large deletions in ITPR1 in SCA15/SCA16 should be performed. As the cost of genetic testing continues to decrease, the complex genetic testing strategies discussed here may be replaced by whole-genome sequencing (WGS), which is not currently used as a routine diagnostic method.
Patients with sporadic ataxia without a family history of Spinocerebellar ataxias (SCAs) may have a hereditary origin due to spontaneous mutations, decreased penetrance of mutated genes, or errors in the reported pedigree. If acquired causes have been excluded by thorough medical history, laboratory testing including MRI and nerve conduction velocity, then appropriate genetic testing must be performed. Gene screenings conducted on patients with sporadic adult-onset ataxia in the U.S. and Europe revealed that 15%-24% of affected individuals had genetic mutations related to SCAs. SCA6, the most commonly missed form of polyglutamine SCA, is often delayed in diagnosis given its nature of late-onset. The transmitting parent carrying SCA6 gene may have died before the onset of the child's symptoms. Genetic testing strategies may also be applied to sporadic ataxia patients. Friedreich's ataxia, the most common autosomal recessive inherited ataxia, although caused by a trinucleotide repeat similar to SCAs, can mimic sporadic ataxia but can be easily distinguished using dynamic gene testing.
Animal Model
In 2015, a team constructed a human SCA3 knock-in mouse model by replacing the mouse Atxn3 gene's exon 8-11 and 3'UTR regions with those of human mutated ATXN3 cDNA containing 91 CAG repeats. The resulting model, named SCA3-Ki91, showed expression of mutant ATXN3 protein and formation of nuclear inclusions in the whole brain tissue of 12-month-old mice, along with cerebellar Purkinje cell degenerative changes and increased astrocyte proliferation. The SCA3-Ki91 mouse model also displayed CAG repeat instability in mouse germ cells during inheritance.
Another team created a heterozygous Atxn3Q82 mouse model by inserting 82 CAG repeat fragments into the exon 10 CAG repeat region of the mouse genome. The pathology of this model was similar to that of the SCA3-Ki91 mouse model. A large number of nuclear inclusions of mutant ATXN3 protein were found in neurons of the mice, and exhibited tissue-specific expression.
Later, another team constructed an SCA3BAC transgenic mouse model using bacterial artificial chromosome (BAC) carrying the entire human mutated ATXN3 gene, including all exons, introns, up-stream promoters and other regulatory elements. At seven months of age, the brain tissue of the first generation males of the SCA3BAC model was investigated using immunofluorescence analysis and weighed. The weight of the SCA3BAC A male was lower than that of the same aged females in the same cage, indicating severe weight loss and brain atrophy. The red boxed area shows the cerebellum tissue of the mouse and the immunofluorescence analysis results revealed significant aggregation of ATXN3 protein in the cerebellum, brainstem, and hippocampal cortex in the SCA3BAC model at seven months compared to wild-type mice, with the cerebellum showing the most significant aggregation. However, no significant ATXN3 was detected in the cortex and striatum. The brain regions most affected in SCA3 patients were cerebellum and brainstem, with less involvement in the cortex and striatum. The results suggest that the affected brain regions in the SCA3BAC model are more consistent with those in SCA3 patients[2].
Table 2. Comparison of body weight and brain tissue weight between BAC-SCA3 first generation mice and wild-type mice at different ages [2].

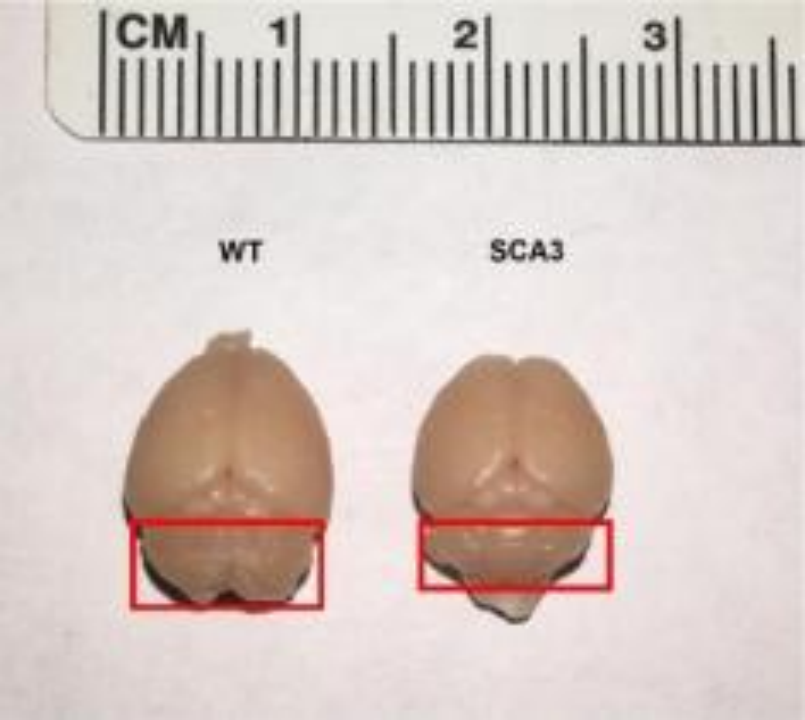
Figure 5. Comparison of brain volume between 7-month-old BAC-SCA3 first generation mouse A and wild-type mouse [2].
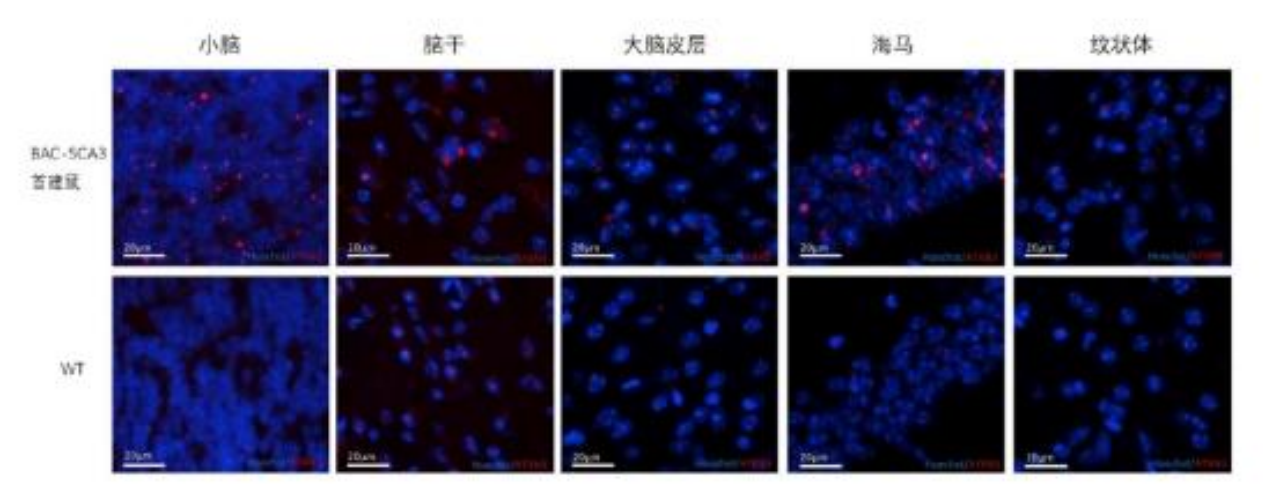
Figure 6. Aggregation of ATXN3 protein in different brain regions of BAC-SCA3 first generation mouse A and wild-type mouse. Blue fluorescence indicates nuclear staining, and red fluorescence indicates ATXN3 protein. The top images show BAC-SCA3 first generation mouse A, and the bottom images show the wild-type mouse [2].
Treatment Options
Currently, there is no effective treatment available for SCA, and symptomatic supportive care is the mainstay of clinical management. In recent years, with the in-depth research of the pathophysiological mechanisms underlying SCA, new therapeutic targets such as antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs) have been discovered in clinical settings, but none have entered clinical trials [1].
Pharmacological treatments: Currently, no drugs have been approved for routine treatment of SCA. For ataxic symptoms, several small studies have examined various drugs, including: 1) Antioxidants: Coenzyme Q10, vitamin E, idebenone, etc; 2) Neurotrophic agents: Adenosine, creatine, B vitamins, etc; 3) Sympathomimetic suppressants: Amphetamines, modafinil, etc; 4) Metabolism-improving therapy: Piracetam, levocarnitine, iron chelators, donepezil hydrochloride, etc; 5) Other drugs: Recombinant human erythropoietin, potassium channel blockers, L-serine, etc. [3].
Supportive care: 1) Physical therapy: Physical therapy can be performed in the early stages of SCA to develop strategies for maintaining normal functions (limb balance, coordination, normal posture) and prevent falling; 2) Occupational therapy: When daily activities become increasingly difficult, it is recommended to involve an occupational therapist to intervene. The intervention should focus on meeting the functional goals and occupational needs of the patients and their caregivers; 3) Speech and speech therapy: Speech therapists provide treatment based on the condition, using acoustic instruments, guidance, and repetitive exercises or visual and auditory aids.
Gene therapy: Gene therapy aims to reduce the levels of mutant-expanded polyQ protein expression by different methods, including the use of antisense oligonucleotides (ASOs), DNA editing technology CRISPR/CRISPR-Cas9, or RNA interference to knock out the polyQ gene. By injecting ASOs into the brain ventricles of SCA2 mouse models, SCA2 model mice showed improved motor symptoms and decreased ATXN2 expression in the cerebellum, leading to reduced cerebellar ataxin-2 protein levels. In addition to ASOs, RNA interference can also be used to suppress the expression of pathogenic genes, and studies have shown that RNA interference has improved motor function in SCA3 mouse models by suppressing ATXN3. CRISPR/Cas9 may have potential in clinical trials of polyQ SCAs, but some issues such as genome instability still need to be resolved. Currently, various gene therapy methods for polyQ SCAs are still in preclinical trials. With the deepening research, gene therapy has the potential to treat polyQ SCAs from the perspective of pathogenesis and to modify diseases for various subtypes of SCAs [4].
Stem cell transplant therapy: Gene therapy is still in the research stage, with most methods being the selective inhibition of SCAs pathogenic gene expression. Stem cell transplantation has been performed in domestic clinical settings, wherein the basic principle is to induce pluripotent stem cells to differentiate into neural stem cells, replace damaged cells, and provide a better microenvironment for neuroconduction through paracrine effects. However, its effectiveness and safety still need to be further observed and studied [3].
Neurostimulation therapy: Neurostimulation therapy mainly includes deep brain stimulation (DBS), transcranial magnetic stimulation (TMS), and transcranial direct current stimulation (tDCS). DBS requires invasive electrode installation through surgery, while the rest are non-invasive. In recent years, multiple studies have shown that neurostimulation may be helpful in improving ataxia symptoms [4].
This article is a collection and summary of information. If there are any errors or omissions, please feel free to provide criticism and corrections!
Reference sources and image sources:
[1] Wu, F., & Zhong, M. (2021). Clinical manifestations, pathogenesis and diagnosis and treatment of spinocerebellar ataxia. Shandong Medical Journal, 61(23), 112-115.
[2] Du, Y. (2020). Clinical research of Chinese Spinocerebellar ataxia type 3 and construction of a novel BAC-SCA3 transgenic mouse model (Doctoral dissertation). Zhejiang University. DOI: 10.27461/d.cnki.gzjdx.2020.002464.
[3] Wang, D., Huang, Y., & Yang, B. (2016). Gene diagnosis and treatment of spinocerebellar ataxia. Journal of Xinxiang Medical College, 33(07), 639-641.
[4] Sun, D., & Hu, X. (2021). Research advances in the treatment of polyglutamine spinocerebellar ataxia. General Medical Clinical and Educational Journal, 19(04), 351-354. DOI:10.13558/j.cnki.issn1672-3686.2021.004.020.