

遗传性小脑共济失调(SCA)[2]——机理研究
疾病诊断
在对一个进行性共济失调患者进行遗传性脊髓小脑共济失调 (spino cerebellar ataxia,SCA) 诊断之前,应通过详细的病史询问、体格检查、实验室检查等除外获得性的病因,因SCA的临床症状有很大的重叠性及异质性,故确诊主要依靠基因检测。SCA的诊断依据如下:①以进行性共济失调为主要临床表现,并除外获得性病因导致的共济失调;②上一代有类似疾病的病史;③SCA基因检测呈阳性[1]。
如果诊断SCA的临床证据明确,则应启动分子遗传学检测。只有在特殊情况下,如已知家族中特定的SCA基因型、临床表型高度提示某一种SCA (例如SCA7中的视力丧失) 或某一种SCA的地区流行率较高 (例如古巴的SCA2),才建议进行有针对性的单一基因检测。而在其他情况下,多推荐采用系统的SCA基因检测。首先要进行多聚谷酰胺SCA的动态突变检测 (即SCA1、SCA2、SCA3、SCA6、SCA7、SCA17、DRPLA),因为此类SCA患病率相对较高。当多聚谷酰胺的CAG动态突变检测为阴性时,可根据现实情况选择适宜顺序:①全外显子测序,可检测传统突变类型的SCA,也可以在发现新的共济失调基因时进行重新分析;②WES不能检测到重复突变,为了评估SCA的所有遗传原因,则必须对非翻译区的重复突变(即SCA8、SCA10、SCA12、SCA31、SCA36和SCA37)进行动态突变的检测;③如果全外显子测序未包含对于具有特定缺失的搜索,则需对SCA15/SCA16的ITPR1中大的缺失进行特定检测。由于基因检测的成本在不断下降,这里讨论的复杂基因测试策略很可能会被全基因组测序所取代,但目前全基因组测序还不能作为常规检测手段。
没有SCA家族史的共济失调患者,也可能由于自发突变、突变外显性降低或错误的亲子关系而有遗传性病因,如通过仔细的病史询问、实验室检查,包括MRI和神经速度传导等,已除外获得性病因的患者则应该进行合理的基因检测。对美国和欧洲的散发性成人型共济失调患基因筛查研究发现,15%-24%的患者SCA相关基因发生突变。SCA6是最易漏诊的多聚谷氨酰胺SCA,因为SCA6是迟发性SCA最常见的类型,传递SCA6基因的亲代可能在患者发病前就已经死亡。对于散发性共济失调患者,以上基因检测策略同样适用,而Friedreich共济失调作为最常见的常染色体隐性遗传共济失调,也可表现为散发性共济失调,虽然同样是由三核苷酸重复造成的,但很容易通过动态基因检测鉴别。
动物模型
2015年有团队构建了人SCA3敲入小鼠模型,用人突变ATXN3cDNA的8-11外显子编码区及3’UTR区域替换鼠Atxn3基因组DNA的8-11号外显子及3’UTR区域,人突变ATXN3带有91个CAG重复序列,该小鼠模型为SCA3-Ki91模型在SCA3-Ki91模型中,他们发现突变ATXN3蛋白在12月月龄的SCA3-Ki91小鼠模型的全脑组织均有表达,并且形成核内包涵体,小脑浦肯野细胞退行性改变伴有星形胶质细胞增生。同时CAG重复序列的传代不稳定性在SCA3-Ki91小鼠模型中也有体现。
另一个团队在小鼠10号外显子CAG重复序列区域插入82个CAG重复片段,构建了杂合Atxn3Q82小鼠,小鼠模型的病理发现和SCA3-Ki91类似,在小鼠的神经元内发现大量的突变ATXN3蛋白的核内包涵体,并且呈组织特异性。
后来又有团队用细菌人工染色体 (Bacteria artificial chromosome, BAC) 构建SCA3BAC转基因小鼠模型,带有全长的人突变ATXN3基因,包括全部外显子、内含子,以及上游的启动子和其他调控元件。在BAC-SCA3首建鼠到达7个月月龄时,它的全脑组织进行重量检测和免疫荧光检测。BAC-SCA3雄性首建鼠A的体重和脑组织重量都比同笼同月龄的雌鼠小,说明该首建鼠出现了严重的体重下降和脑萎缩。红色方框里面显示的是小鼠的小脑组织;在免疫荧光检测结果可以看出BAC-SCA3小鼠在7个月时,与野生型小鼠相比,小脑、脑干和海马皮层有显著的ATXN3蛋白聚集现象,以小脑更为显著,然而在大脑皮层和基底节并未检测到明显的ATXN3。SCA3患者受累脑区也以小脑和脑干为主,皮层和基底节受累较少。该结果说明BAC-SCA3小鼠的受累脑区和SCA3患者的受累脑区较为一致[2]。
表1 月龄BAC-SCA3首建小鼠与野生型小鼠体重及脑组织重量的比较[2]

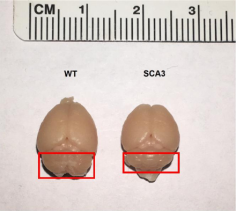
图1 7月龄BAC-SCA3首建小鼠A与野生型小鼠的脑体积大小的比较[2]
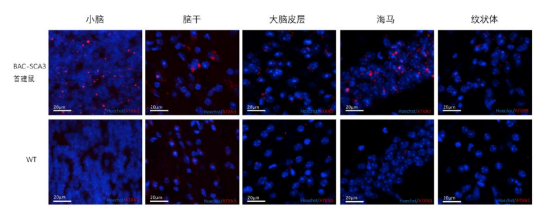
图2 BAC-SCA3首建鼠A和野生型小鼠不同脑区ATXN3蛋白的聚集。
蓝色荧光为细胞核染色,红色荧光为ATXN3蛋白。上为BAC-SCA首建鼠A,下组为野生型小鼠[2]
治疗方式
目前SCA缺乏有效的治疗方法,临床上主要以对症支持治疗为主。随着近年来对SCA病理生理机制研究的不断深入,临床发现了反义寡核苷酸(ASO)、小干扰RNA(siRNA)等新的治疗靶点,但均未进入临床试验[1]。
药物治疗:目前还没有药物被批准用于SCA的常规治疗。对于共济失调症状,一些小型研究已经对多种药物进行了研究。(1)抗氧化剂:辅酶Q10和维生素E、艾地苯醌等。(2)神经营养药物:三磷腺苷、肌酐、维生素B族等。(3)交感神经药物抑制剂:安非他明、莫达非尼等。(4)改善代谢治疗:吡拉西坦、左旋肉碱、铁螯合剂、盐酸多奈哌齐等。(5)其他药物:人促红细胞生成素、钾通道阻滞剂、环丝氨酸等[3]。
支持治疗:①物理治疗:SCA患者发病早期即可进行物理治疗,以便早期制定维持正常功能 (肢体平衡、协调、正常姿势) 的策略和防止跌倒。②职业治疗:当患者日常活动越来越困难时,推荐职业治疗师的介入,干预应侧重于满足患者及照顾者的功能目标和职业需求。③语言及言语治疗:语言治疗师根据病情通过声学仪器、指引及重复性练习,或利用视觉及听觉辅助等方法给予治疗。
基因治疗:基因治疗是通过各种方式减少变异扩增的polyQ蛋白表达水平,主要方法包括运用反义寡核苷酸(anti sense oligonucleotide, ASOs),DNA编辑技术CRISPR/CRISPR-Cas9或是通过RNA干扰敲除改变polyQ基因。同样ASOs经脑室注射入不同的SCA2模型小鼠,发现SCA2小鼠的运动症状有改善并且降低了小脑中ATXN2表达,小脑ataxin-2蛋白减低。除了运用ASOs,还可以通过RNA干扰抑制致病基因的表达,有研究用RNA干扰抑制在SCA3小鼠模型中的ATXN3,显示可以提高其运动功能。CRISPR/Cas9可能在polyQ SCAs的临床试验中具有潜力,但是包括基因组不稳定等问题仍需要解决。现阶段运用各种基因治疗polyQ SCAs的方法都在临床前试验阶段,随着研究的不断深入,基因治疗有潜力从病因方面治疗polyQ SCAs,并对各亚型SCAs进行疾病修饰治疗[4]。
干细胞移植治疗:基因治疗尚在研究阶段,原理多为选择性抑制SCAs致病基因表达。干细胞移植手术已在国内临床上相继开展,其基本原理为诱导多能干细胞分化为神经干细胞,替换受损细胞,并通过旁分泌作用为神经传导提供更好的微环境。但其有效性和安全性还有待于进一步观察研究[3]。
神经调节治疗:神经调节治疗主要包括深部脑刺激(deep brain stimulation, DBS),经颅磁刺激 (transcranial magnetic stimulation, TMS) 以及经颅直流电刺激 (transcranial direct current stimulation, tDCS),除了DBS需要经手术有创安装电极,其余均是无创的。近年来,多项研究显示神经调节可能对改善共济失调的症状有帮助[4]。
了解更多
想了解遗传性脊髓小脑共济失调相关基因及小鼠模型的更多信息,上RDDC官网,即可免费查看哟!
声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考文献及图片来源:
[1]吴方瑞,钟敏.脊髓小脑性共济失调的临床表现、发病机制及诊疗研究进展[J].山东医药,2021,61(23):112-115.
[2]杜依楚. 中国人群脊髓小脑性共济失调3型的临床研究及新型BAC-SCA3转基因小鼠模型的构建[D].浙江大学,2020.DOI:10.27461/d.cnki.gzjdx.2020.002464.
[3]王东浩,黄艳梅,杨保胜.脊髓小脑性共济失调的基因诊断及治疗研究进展[J].新乡医学院学报,2016,33(07):639-641.
[4]孙迪,胡兴越.多聚谷氨酰胺脊髓小脑性共济失调的治疗研究进展[J].全科医学临床与教育,2021,19(04):351-354.DOI:10.13558/j.cnki.issn1672-3686.2021.004.020.