

苯丙酮尿症(PKU)——"不食人间烟火的天使"
疾病概述
我们身边有一些人,除了极少数的蔬菜水果可以吃以外,其他的食物他们都不能吃,他们这一生只能吃一些特别的米和面,其他的营养都来源是一种特别的“奶粉”,他们就是苯丙酮尿症患者。

图1 苯丙酮尿症患儿只能吃特定食物 图源来自网络
苯丙酮尿症 (PKU) 是一种常染色体隐性遗传病,属于苯丙氨酸羟化酶 (PAH) 缺乏症之一,是PAH基因发生了错义突变导致苯丙氨酸羟化酶活性降低或丧失,影响了苯丙氨酸 (Phe) 在体内的正常代谢,使苯丙氨酸 (Phe) 不能转化为酪氨酸 (tyrosine,Tyr) ,酪氨酸及其他正常代谢产物合成减少,血液中Phe含量增加,影响中枢神经系统发育。同时次要代谢途径增强,生成苯丙酮酸、苯乙酸和苯乳酸,并从尿中大量排出,苯乳酸使患儿的尿液具有特殊的鼠尿臭味。苯丙酮尿症是高苯丙氨酸血症的主要类型[1,5]。
流行病学
PKU发病率在性别上无统计学差异,但突变种类和频率存在明显地域和种族差异。美国白种人新生儿PKU发病率为1/10000,土耳其新生儿PKU发病率为1/2600,为全世界最高;芬兰和日本的新生儿PKU发病率最低,不足1/100000。1985-2018年中国大陆研究显示PKU发病率为1/11144~11614,且呈现“北高南低”趋势。另外,欧美白种人PKU常见突变位点位于外显子12和内含子12,而中国大陆人群常见突变位点位于外显子7、外显子6和外显子12。可见PKU致病基因PAH的高度异质性[2]。
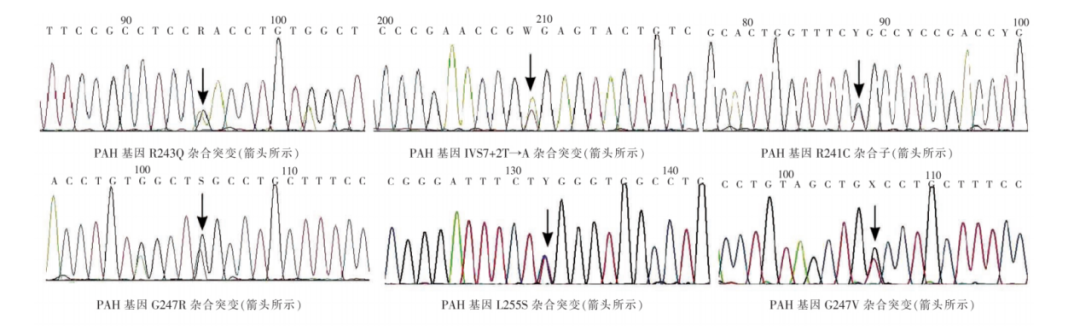
图2 PAH基因外显子7位点突变基因型[2]
近期有学者在吉林1个家系样本中发现了新的突变位点F294S,该位点之前未见报道过,其余突变位点均已被收录在PAH Mutation Analysis Consortium Database (HGMD)数据库中。依据ACMG发布的《遗传变异分类标准与指南》进行评估。该突变点符合3个中等 (PM1、PM2、PM3) 和1个支持 (PP3) 证据,依据指南,目前致病性评估为可能致病 (likely pathogenic,LP) [3]。
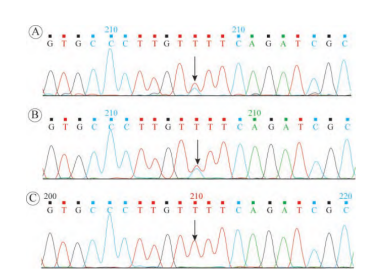
图3 F294S 一代测序验证结果
A:患儿F294S电泳结果:Exon8 c.881T>C, p.F294S,杂合突变;B:患儿父亲相同位点一代验证电泳结果:Exon8 c.881T>C, p.F294s,杂合突变;C:患儿母亲相同位点一代验证电泳结果,为野生型[3]
疾病分类
PKU临床分类比较复杂,依据血苯丙氨酸浓度、对四氢生物蝶呤反应性、苯丙氨酸耐受性、临床症状、苯丙氨酸负荷试验、PKU表型等指标和目的有不同的分类方式。其中根据患者能否对四氢生物蝶呤的治疗产生反应将PKU分为2类:一类患者对四氢生物蝶呤的治疗产生反应,包括对该治疗产生反应的苯丙氨酸羟化酶缺陷型PKU和四氢生物蝶呤缺乏型PKU;另一类则是对该治疗不产生反应的苯丙氨酸羟化酶缺陷型PKU;也有学者依据苯丙酮尿症患者治疗前血液中苯丙氨酸浓度大小,将苯丙酮尿症分为经典PKU (classic phenylketonuria) 、中度PKU (moderate phenylketonuria) 、轻度PKU (mild phenylketonuria) 和MHP (mild hyperphenylalaninemia) 4类,其中经典PKU患者、中度PKU患者体内苯丙氨酸浓度分别为大于1200μmol/L和900~1200μmol/L;轻度PKU患者和MHP患者体内苯丙氨酸浓度为600~900μmol/L和120~600μmol/L[4]。
临床表现
新生儿期的苯丙酮尿症患儿无任何临床表现,出生3个月后可出现智力发育迟缓和语言发育落后等典型症状,并随着年龄增大病情逐渐加重,1岁时症状明显。患儿有不同程度的智力低下,60%属重度低下 (IQ低于50) ,出生数月后因黑色素合成不足,其毛发和虹膜色泽逐渐变浅,为黄色或棕黄色,皮肤白。由于尿液、汗液含有大量苯乳酸而有鼠尿臭味。随着年龄增长,逐渐表现出智能发育迟缓,以认知发育障碍为主、小头畸形、癫病发作,也可出现行为、性格等异常,如多动、自闭症、自卑、忧郁等神经系统表现。婴儿期还常出现呕吐、湿疹等。治疗越晚,智能损害越严重[6]。
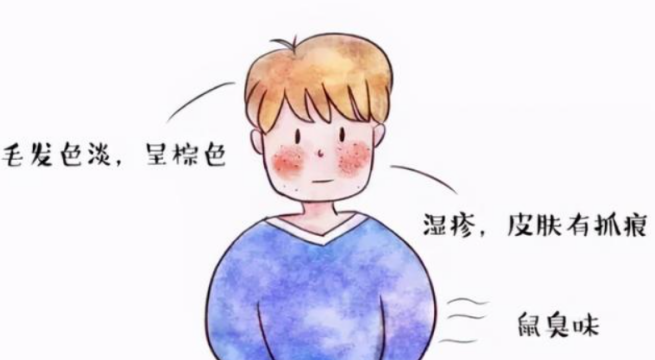
图4 苯丙酮尿症临床表现 图片来源于网络
疾病诊断
1.新生儿期筛查和诊断:采取新生儿喂奶72小时足跟血,滴于滤纸片上,晾干即可寄往筛查实验室。目前主要采用Guthrie细菌抑制法测定血斑苯丙氨酸浓度。初筛阳性者进一步用HPIC进行苯丙氨酸浓度测定,一般正常新生儿血液苯丙氨酸含量上限为120 μmol/L (2mg/dL) ,未经治疗的典型患儿血浆苯丙氨酸可高达240 μmol/L (401mg/dL) 以上。
2.四氢生物蝶呤 (BH4) 负荷试验:经典型PKU患儿血苯丙氨酸浓度在服用BH4,前后无大改变,非经典型PKU患儿在服用4小时后血浆苯丙氨酸即明显下降。由于非经典型PKU患儿的神经系统损害严重,且单纯饮食治疗效果不佳,故对每一例高苯丙氨酸血症患儿均应进行BH4负荷试验,以早期鉴别,采必要的治疗措施。应用HPLC测定尿液中新蝶呤和生物蝶呤的含量,可以鉴别各型PKU3. 基因诊断PAH基因的DNA突变分析可用于预测PAH缺陷的生化表型。突变分析也是.进行携带者诊断和产前诊断的技术基础。基于PCR扩增的直接分子诊断方法或单倍型分析间接诊断方法是目前应用于PAH基因检测的主流技术。
3.有学者提出辅助诊断方法——双酶级联反应法检测苯丙酮尿症:血液中苯丙氨酸的量对新生儿苯丙酮尿症的诊断具有重要意义。本工作以NADH为检测的“信使”,串联使用硝基还原酶和苯丙氨酸脱氢酶的催化反应实现了对苯丙氨酸简单高效的检测。检测限达0.9ppm,对常见的苯丙氨酸干扰物有很好的抗干扰能力[7]。
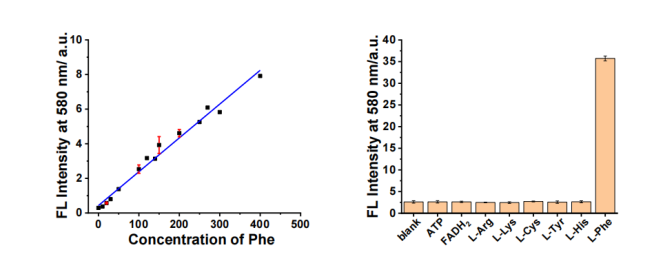
图5 传感平台对苯丙氨酸浓度的荧光响应(左)与传感平台对不同物种的荧光活性的线性关系图(λex=540 nm)[7]
致病机理
①经典型苯丙酮尿症 (PKU) 。
正常情况下,苯丙氨酸是通过肝细胞产生的苯丙氨酸羟化酶 (PAH) 转化为酪氨酸,之后再分别合成甲状腺素,肾上腺素和黑色素等途径来完成代谢(如图1)。
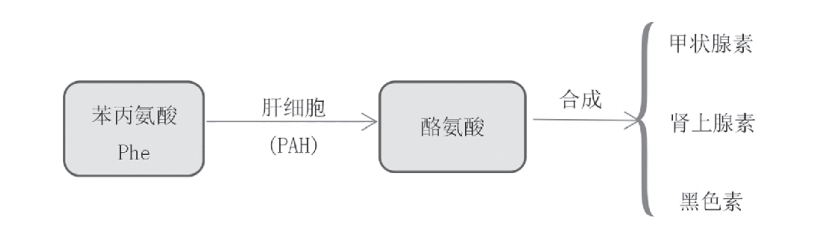
图1 正常人体内苯丙氨酸代谢途径[8]
而PKU患者由于缺乏苯丙氨酸羟化酶,使得苯丙氨酸不能羟化为酪氨酸,所以遵循另一条代谢通路,即苯丙氨酸 (Phe) 与α-酮戊二酸进行转氨基作用,生成苯丙酮酸,引发大量的苯丙酮酸在血液和组织中堆积,最后于尿液中排出。而苯丙酮酸的代谢产物在中枢神经积累并产生毒性,会导致相应的神经系统症状(如图2)。
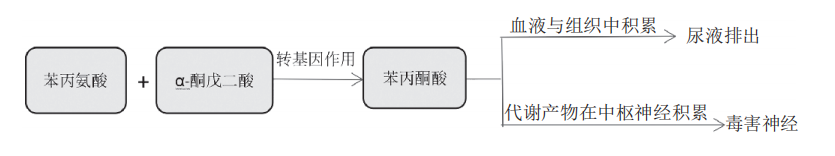
图2 PKU患者体内的苯丙氨酸代谢途径[8]
②四氢生物蝶呤 (BH4) 缺乏症。
三磷酸鸟苷 (GTP) 在三种合成酶作用下,合成无活性的四氢生物蝶呤,后者与氧气结合生成4α-羟化四氢生物蝶呤,然后在翼素-4α-卡宾明脱水酶 (PCD) 作用后生成 (二氢生物蝶呤) BH2,再在二氢生物喋呤还原酶 (DHPR) 作用下,生成具有生物活性的BH4,发挥重要的生理作用 (如图3)。
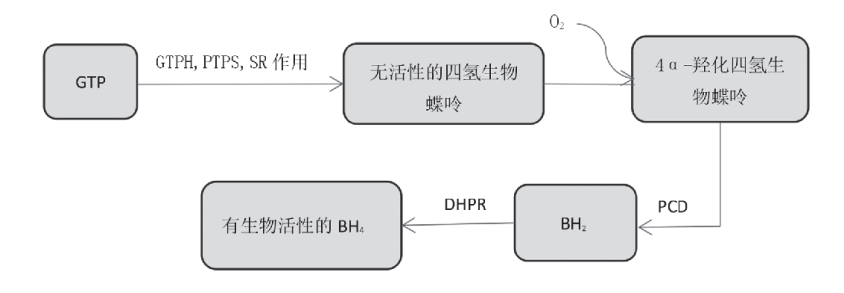
图3 四氢生物喋呤生成过程[8]
生成的BH4会与Phe通过一种GTPCH刺激蛋白 (P35) 起着调节GTPCH的作用,当血Phe增高时,通过增强GTPCH作用促进新喋呤和生物喋呤的合成 (如图4)[1]。
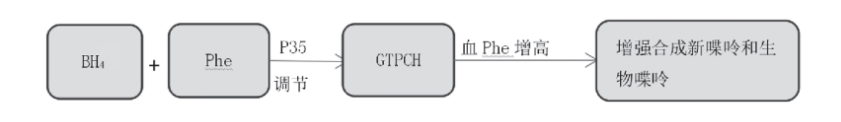
图4 四氢生物喋呤作用过程[8]
小鼠模型
目前应用广泛的苯丙酮尿症疾病小鼠模型为Pahenu1和Pahenu2两种,这两种模型小鼠都是用化学诱变剂乙酰基亚硝基脲 (ENU) 诱变获得。但Pahenu2小鼠肝脏所表达的突变蛋白基本上检测不到催化活性,此外Pahenu2小鼠在7号外显子上发生点突变,携带这一突变型的患者比Pahenu1小鼠突变型携带者更多。因此对于苯丙酮尿症的研究使用的多是Pahenu2模型小鼠。
还有学者利用CRISPR/Cas9系统构建丙酮症疾病小鼠模型PahR408W。根据野生型小鼠Pah基因的12号外显子序列设计靶点并设计同源重组模板,同源重组模板上带有胞嘧啶突变成胸腺嘧啶的点突变,从而在DNA修复过程中在小鼠胚胎DNA上引入胞嘧啶到胸腺嘧啶的点突变,最终将PAH蛋白氨基酸序列408位的精氨酸突变成色氨酸 (图5) 。通过显微注射的方式引入CRISPR/Cas9元件,小鼠出生后检测F0代小鼠基因型,最终获得阳性小鼠25只,其中PahR408W小鼠3只。且PahR408W小鼠具有典型的苯丙酮尿症病症,正常生理状况下,体内的苯丙氨酸在苯丙氨酸羟化酶作用下代谢生成酪氨酸,随后一部分酪氨酸在相关酶的作用下生成黑色素。当体内苯丙氨酸羟化酶活性降低,不能正常代谢苯丙氨酸成酪氨酸,因此不能合成足够多的黑色素,导致出现毛色色素衰退的症状。首先对成年 (8周龄) 野生型和PahR408W小鼠进行毛色对比。通过与野生型小鼠对比,发现成年 (8周龄) PahR408W小鼠毛色出现明显的色素减退的症状 (图6)[9]。
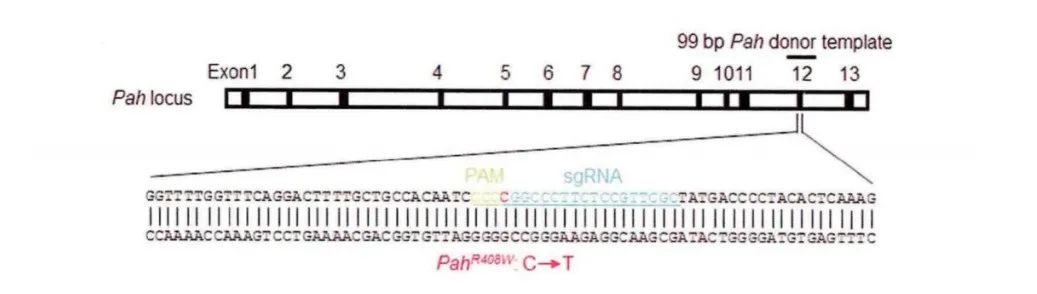
图5 PahR408W模型设计示意图。蓝色碱基为靶点sgRNA序列,红色碱基为突变前的碱基,绿色碱基标注出PAM区域[9]

图6 小鼠毛发颜色图[9]
治疗方法
目前尚无方法可以根治PKU,最常见的方法是在新生儿筛查中做到“早发现早治疗”,减少对患儿生长发育及智力发育等方面损害。因此快速、有效、精准的基因诊断对PKU患儿的尽早治疗十分有意义[10]。当前PKU治疗方法主要以饮食疗法、糖巨肽法、大中性氨基酸法、补充四氢生物蝶呤法为主,其他潜在疗法包括酶替代疗法和基因疗法仍在探究中。
饮食疗法
如果PKU患者不进食或少进食含苯丙氨酸的食物,则PKU基因未必会对患者造成伤害,所以限制苯丙氨酸摄入不失为一个好方法。由于大部分食物中都含有苯丙氨酸,低苯丙氨酸饮食来源有两种,一种是天然低蛋白食物,另一种为人工合成的氨基酸药食。对于纯天然的低蛋白食物仍然要严格控制摄入量,且要补充其他人体必要氨基酸,维生素等,以免造成营养失衡和骨质疏松、神经损伤加剧等并发症,而人工合成的氨基酸药食口味差,进食量又大,会导致患者生活质量下降,对饮食疗法的遵循度降低[11]。
糖巨肽疗法 (glycomacropeptides, GMP)
GMP是来自源于干酪乳清的天然蛋白质,其苯丙氨酸含量低,但富含缬氨酸、异亮氨酸和苏氨酸等人体必需氨基酸,也可作为患者低苯丙氨酸食物的来源,目前已用于PKU的治疗。GMP饮食疗法可与其他疗法联合使用,用于治疗PKU患者尤其是需严格控制苯丙氨酸摄入的患者。然而该疗法具有免疫性不明确且缺乏长期食用的临床安全性数据等缺点,该方法用于PKU的治疗仍需进一步观测。
大中性氨基酸法 (large neutral amino acids, LNAA)
大中性氨基酸包括支链氨基酸 (缬氨酸、亮氨酸、异亮氨基酸) 和芳香氨基酸 (苯丙氨酸、酪氨酸) 等,这些大中性氨基酸可与苯丙氨酸竞争性与脑部L型氨基酸运载体结合,抑制苯丙氨酸通过胃肠黏膜和血脑屏障,进而降低苯丙氨酸在胃肠道的吸收和减少其进入脑部的量。故通过饮食增加血液中其他LNAA的浓度,也成为治疗PKU的一种方法。但该方法一般推荐成人患者使用。该方法缺乏长期临床试验数据,需进一步研究来证明其治疗结果的安全性和有效性。
补充四氢生物蝶呤疗法
四氢生物蝶呤作为苯丙氨酸羟化酶的辅助因子,能够显著降低BH4缺乏型和对BH4治疗产生反应的PAH缺乏型患者体内苯丙氨酸浓度。二盐酸沙丙蝶呤 (sapropterin dihydrochloride) 是人工合成的BH4,最初是由日本Daiichi Sankyo公司研制,其片剂由BioMarin Internation公司研制,商品名:KUVAN。该化合物于2007年被美国FDA批准上市,用于治疗苯丙酮尿症及高苯丙氨酸血症。
苯丙氨酸氨解酶法 (phenylalanine ammonialyase, PAL)
研究发现,通过注入外源性苯丙氨酸氨解酶,可降低患者体内苯丙氨酸浓度,从而达到治疗疾病的目的。因为苯丙氨酸氨解酶可将苯丙氨酸降解为E-肉桂酸和少量的氨,E-肉桂酸可迅速转化为马尿酸,随尿液排出体外,所以此代谢途径可降低患者体内苯丙氨酸浓度。目前,已经启动PEG-PAL的Ⅲ期临床研究,用于评价药物的安全性和有效性[12]。
基因疗法
近年来随着生物医药技术的发展,基因疗法取得了很大的进展。研究发现,98%的PKU患者是由于苯丙氨酸羟化酶基因突变导致患者体内苯丙氨酸羟化酶缺乏或活性不足。苯丙氨酸羟化酶致病突变基因大多位于外显子区域,且主要集中在外显子3、外显子6、外显子7和外显子11上。通过基因转移方法,将含有鼠源PAH-cDNA注入到PKU模型小鼠体内,可恢复小鼠肝脏中苯丙氨酸羟化酶的活性,使苯丙氨酸的代谢恢复正常,从而降低小鼠体内苯丙氨酸的浓度。另一种研究方法是基于PKU患者体内苯丙氨酸羟化酶基因的无义突变。大约10% 的PKU患者是由于苯丙氨酸羟化酶基因发生无义突变,产生不稳定或者无活性的截短蛋白。对携带PAH基因无义突变的哺乳动物细胞株 (COS-7和HEK293) 给予氨基糖苷类药物庆大霉素和遗传霉素 (geneticin,G418) ,该细胞株在体外可表达全长并且有活性的PAH。结果显示,无义突变药物也是一种潜在治疗PKU疾病的方法,但仍然需要对其进行体内药效学研究[4]。
参考文献以及图片来源:
[1]何江,强荣,毛新梅,王慧珍,闫有圣,邹红云,史清海.中国西北地区汉族苯丙酮尿症患者苯丙氨酸羟化酶基因突变分析[J].中国实验诊断学,2022,26(03):317-322.
[2]周怡,阙敏,王新静.苯丙酮尿症患儿苯丙氨酸羟化酶基因外显子7突变特点及临床研究[J].川北医学院学报,2021,36(10):1338-1341.
[3]李锦,李晶,李娜娜,陈宁,孟岩,潘亭亭,张蔓丽,朱军,田亚平.吉林和辽宁地区80例苯丙酮尿症患儿苯丙氨酸羟化酶基因突变分析[J].解放军医学院学报,2021,42(08):843-848+889.
[4]牛瑞青,冯文化.苯丙酮尿症及相关治疗方法研究进展[J].中国新药杂志,2018,27(02):154-158.
[5]邵婷婷. 以苯丙酮尿症小鼠为模型探索基因编辑修复遗传突变的策略[D].华东师范大学,2019.DOI:10.27149/d.cnki.ghdsu.2019.000332.
[6]张彩芳.早期筛查 防治苯丙酮尿症[J].健康向导,2022,28(01):28-29.
[7]孟磊磊,曾毅,李嫕. 双酶级联反应法检测苯丙酮尿症[C]//.中国化学会第十六届全国光化学学术讨论会会议论文集(2019).[出版者不详],2019:195.DOI:10.26914/c.cnkihy.2019.076975.
[8]朱晓涵.苯丙酮尿症的致病机理与防治措施研究[J].当代化工研究,2018(09):187-189.
[9]马列. CRISPR-Cas9技术在苯丙酮尿症小鼠模型构建及基因治疗的应用研究[D].华东师范大学,2017.
[10]李锦,李晶,李娜娜,陈宁,孟岩,潘亭亭,张蔓丽,朱军,田亚平.吉林和辽宁地区80例苯丙酮尿症患儿苯丙氨酸羟化酶基因突变分析[J].解放军医学院学报,2021,42(08):843-848+889.
[11]朱晓涵.苯丙酮尿症的致病机理与防治措施研究[J].当代化工研究,2018(09):187-189.
[12]马列. CRISPR-Cas9技术在苯丙酮尿症小鼠模型构建及基因治疗的应用研究[D].华东师范大学,2017.