

尼曼-匹克病(NPD)——逆风的“舞蹈”
疾病概述
有这样一种疾病,会导致患者智力逐渐退化到婴儿水平,他们的生活就像是被按了倒退键,一步步逐渐丧失行动能力、语言能力、记忆力,同时还会伴随着肝脾肿大、癫痫抽搐等症状,这就是尼曼-匹克病。

图1 尼曼-匹克病患者末端神经受到严重的破坏,手脚呈畸形状态[4]
尼曼-匹克病(Niemann-Pick disease,NPD)是由尼曼和匹克两人分别于1914年和1922年报道描述,属于先天性糖脂代谢性疾病,是一种罕见的常染色体隐性遗传病,是由于神经鞘磷脂酶的先天性缺陷,致使神经鞘磷脂代谢障碍。神经鞘磷脂是一切细胞的质膜和亚细胞器的脂质成份,也是红细胞基质和神经髓鞘的主要脂质组分,因而,此病累及全身。神经鞘磷脂在细胞代谢衰老过程中被巨噬细胞吞噬后,经神经鞘磷脂酶将神经鞘磷脂降解成N-酰基氨基醇及磷酸胆碱,此酶存在于肝、脾、肾、脑和小肠。当此酶活力降低至正常的50%以下,不能将不断产生的神经鞘磷脂降解,网状内皮细胞因清除衰老细胞的产物积脂极多,形成含有神经鞘磷脂的泡沫细胞,导致肝、脾肿大和中枢神经系统的退行性变[1]。
发病机制
根据基因突变及发生机制的不同,NPD分为3种类型:(1)A型和B型(NPD-A/B),由溶酶体酶神经鞘磷脂酶缺乏致鞘磷脂沉积所引起;(2)C型(NPD-C),由胆固醇从溶酶体到细胞溶质的转运障碍导致[2]。
NPD-A/B的致病基因SMPD1位于染色体11p15.1-p15.4,含6个外显子,编码含629个氨基酸的糖蛋白。该基因已发现有180多种突变,包括错义突变、无义突变、缺失突变及剪接突变。SMPD1基因编码酸性鞘磷脂酶(acid sphingo myelinase,ASM),ASM缺乏导致其降解的底物鞘磷脂在单核-吞噬细胞系统及脑组织贮积。
NPD-C1位于18q11-a12,含25个外显子,已知突变超过300种,p.l1061T突变最为常见,90%的尼曼-匹克病C型患者是由于NPD-C1基因突变所致。NPD-C2位于14q24.3,含5个外显子,已报道突变位点有30个,4%的尼曼-匹克病C型患者是由于NPD-C2基因突变所致。NPD-C1或NPD-C2基因突变后,胆固醇转运障碍,游离的胆固醇在溶酶体内贮积致病。
国外不同人种之间NPD-A/B型发病率为1/250000-1/44960,NPD-C型发病率为1/150000-1/100000,国内缺乏准确的发病率调查[3]。
临床表现
NPD-A型(急性神经型或婴儿型)占NPD的85%左右,多于婴儿期起病,临床表现严重,是一种急性神经元病变性疾病,中枢神经系统退行性变早期即可出现。患儿在宫内及娩出时大多正常,少数在新生儿期表现为黄疸迁延,出生后数周即可出现肌力和肌张力低下,从而发生喂养困难、体质量不增,并常伴有反复呕吐、腹泻等症状,3~6 个月时常出现淋巴结肿大和肝脾增大。该型患儿病情常迅速进展,神经系统症状出现也较早,6个月时即可出现精神运动发育迟滞,如出现运动发育迟缓、听力视力逐渐减退直至丧失、表情淡漠、惊厥等症状。约50%的患儿眼底可见黄斑部樱桃红斑,部分患儿可见皮肤棕黄色素沉着,最终患儿常极度消瘦,呈恶病质状态,多数在3岁左右因感染而死亡。
NPD-B型(慢性非神经型或内脏型)发病较A型晚,表现为肝脾肿大,通常先出现脾脏增大,然后出现肝脏增大。严重受累者可出现继发于脾功能亢进的血小板减少症,该型病情进展缓慢,常无神经系统的表现。其他全身表现有身材矮小、骨骼成熟延迟、高脂血症、眼部病变(黄斑晕及樱桃红斑)、间质性肺疾病,肺部常呈弥漫性浸润,易发生感染而迁延不愈。NPD-B型智力正常,可带病长期生存。
NPD-C型(慢性神经型)个体差异显著,发病年龄从围产期至成年人(甚至于70岁发病),一般于5~15岁死亡,主要包括内脏(肝、脾、肺)、神经、精神三方面表现。该型在胎儿期即可表现为胎儿水肿或腹水,新生儿期发病多表现为广泛性内脏受累,常出现肝脾肿大、黄疸消退延迟、间质性肺病或伴精神运动发育迟滞和肌张力减低,婴儿期或儿童期发病常先出现肝脾肿大或单纯的脾脏肿大,而后累及神经系统,主要表现为快速眼动异常、共济失调、辨距不良、吞咽困难、构音障碍和痴呆,痴笑猝倒、癫痫发作、听力损害及肌张力障碍,其中垂直性核上性眼肌麻痹(vertical supranuclear gaze palsy,VGSP)是最具特征性的表现[2]。
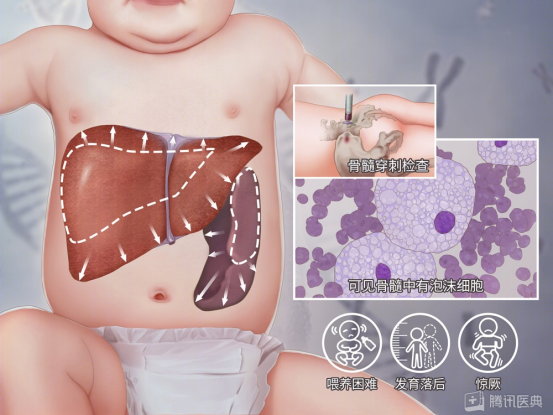
图2 曼尼-匹克病患者症状 图片来源于腾讯医典
辅助检查
NPD-A/B型
1.常规检查
(1)血常规:脾功能亢进患者可出现血小板减少,甚至出现全血细胞减少。
(2)肝功能:大部分患者肝脏转氨酶轻度至中度升高。
(3)血脂:甘油三酯轻中度升高、高密度脂蛋白胆固醇降低、低密度脂蛋白胆固醇升高。
2.酸性鞘磷脂酶活性检测:外周血淋巴细胞或皮肤成纤维细胞培养酸性鞘磷脂酶活性低于正常下限的30%可以确诊NPD-A/B型。
3.影像学检查
(1)肝脏和脾脏:B超、CT或MRI检查可见不同程度的肝大、脾大、肝脾增大或肝硬化表现。
(2)肺部:肺高分辨率CT可以发现小叶间隔增厚,磨玻璃密度影和钙化等。
(3)头颅:NPD-A型患者头颅MRI可以正常,也可以表现为脑萎缩,部分患者可以出现白质T2高信号。
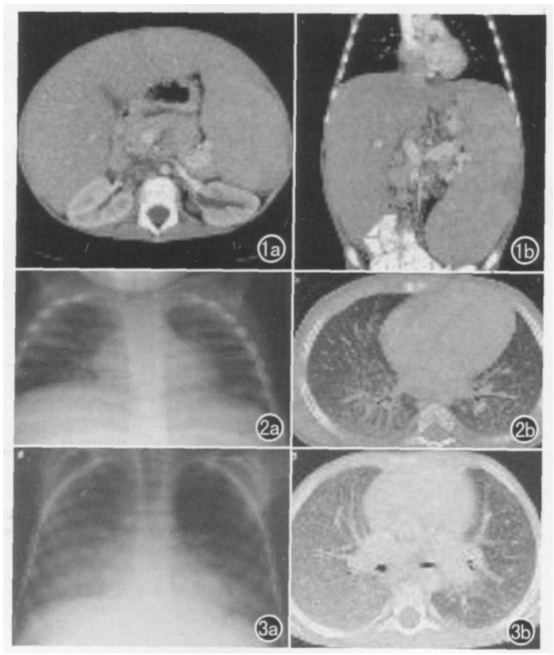
图3 尼曼匹克病B型低剂量MDCT:1a.肾蒂层面轴面扫描;1b.冠状面MPR 均显示明显肝脾肿大巨大脾脏可达小骨盆在侧背旁脾缘处可见楔形梗死区。尼曼匹克病A/B 型(混合型):2a.X线胸片;2b.HRCT 双侧肺网状结节样纹理增多且叶间隔明显增厚。患儿男8岁尼曼匹克病B型:3a.X线胸片;3b.HRCT 大片肺间质性肺部病变尤以右侧为甚伴叶间质增厚和叶间隔增厚以及明显的乳白阴影[5]。
4.组织病理检查:常用组织为骨髓、脾、肝脏、肺及淋巴结。光镜下可以看到富含脂质的巨噬细胞(lipid-laden macrophage),也称泡沫样细胞或尼曼匹克细胞。电镜下泡沫细胞的细胞核小并偏离细胞中心,膜侧因为脂肪蓄积而呈透明状。活组织检查发现泡沫细胞提示尼曼-匹克病可能,但阴性并不能除外此病。
5.SMPD1基因分析:检出2个等位基因已知致病变异可以确诊NPD-A/B型。
NPD-C型
1.常规检查:大多数患者血常规、肝功能等没有明显异常。
2.血浆壳三糖苷酶:壳三糖苷酶由活化的巨噬细胞合成,部分NPD-C型患者血浆壳三糖苷酶活性可有轻度增高,但在晚发型患者该酶不升高。该酶升高还可见于戈谢病、脑卒中及2型糖尿病患者。
3.活组织检查:常用组织为骨髓、脾、肝脏、肺及淋巴结。光镜下可以看到特征性的泡沫细胞。
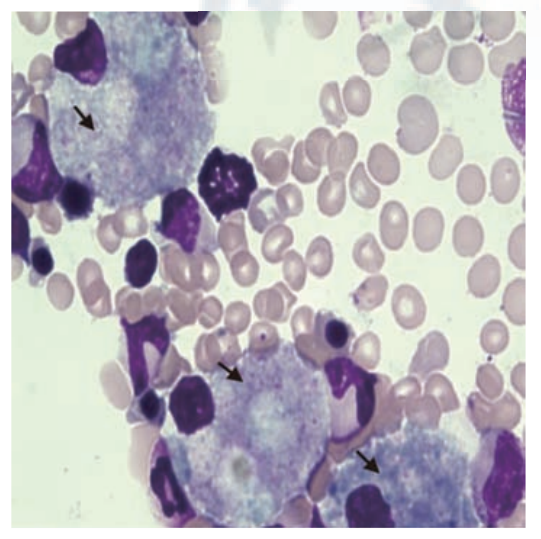
图4 尼曼匹克病C型患者的骨髓涂片 箭头示骨髓中的泡沫细胞(HE染色X1000)[6]
4.成纤维细胞相关检查
(1)Filipin染色:Filipin能与游离的胆固醇特异性结合,荧光显微镜下可见核周溶酶体强荧光信号(即游离胆固醇),为NPD-C阳性细胞,是确诊NPD-C的方法之一。大于80%的NPD-C型病例可以观察到这种典型表现。
(2)胆固醇酯化率的检测:具有经典表型的细胞胆固醇酯化率明显降低甚至为零,而变异型患者的细胞只有轻度的酯化受损。对于这一类患者,基因诊断更加重要。该方法敏感性较Filipin染色低。
5.头颅影像检查:NPD-C型患者的头颅MRI无特异性表现,多数报道提示有小脑、海马、大脑灰质的缩小以及白质的异常信号。
6.基因分析:基因检测可以确诊疾病。对于临床高度怀疑为NPD-C型的患者,即使Filipin染色阴性的患者,均应进行基因分析。NPD-C1或NPD-C2基因检出2个等位基因致病突变有确诊意义[3]。
临床诊断
诊断尼曼-匹克病(NPD)需要依靠详细的病史采集和系统的体格检查,特别关注是否有新生儿期的胆汁淤积、惊厥、猝倒、认知障碍及精神障碍等表现。体格检查包括全身及全面的神经系统和眼科检查,如肝脾、眼球运动、吞咽、肌力、肌张力、反射、步态、小脑体征等。
NPD 临床诊断参照标准:
(1)肝脾肿大;
(2)有或无神经系统损害或眼底樱桃红斑;
(3)外周血淋巴细胞和单核细胞胞浆有空泡;
(4)骨髓可找到泡沫细胞;
(5)X线片肺部呈粟粒样或网状浸润;
(6)有条件者可行神经鞘磷脂酶活性测定、尿神经鞘磷脂排泄量、肝脾淋巴结活检。
目前生化检查均无特异性,NPD的实验室诊断手段除神经鞘磷脂酶活性测定及尿神经鞘磷脂排泄量测定、骨髓涂片、肝/脾或淋巴结组织活检外,还有基因分析,目前国内确诊尼曼-匹克病主要根据临床症状、组织活检及基因分析。
图1 肝脏肿大 图源来自相因网
NPD-A型、B型诊断
若患者淋巴细胞或皮肤成纤维细胞中酸性鞘磷脂酶的活性降低,活性不到正常对照者的10%,则可确定为NPD-A或NPD-B【酸性鞘磷脂酶(ASM)缺乏症】,对于具有典型临床表现的患者,酶检测为首选。另外,因基因检测包可覆盖更广泛的疾病,国外学者建议在有特殊临床表现的溶酶体贮积病中应用基因突变分析帮助确诊该病:通过高通量二代测序检测包括SMPD基因在内的基因检测包,找出SMPD1基因突变的基因位点,然后通过Sanger一代基因测序验证突变基因的位点,再根据临床表现,确定为A型或B型。肝脾大、间质性肺病变、黄斑部樱桃红斑、发育迟缓等临床症状提示NPD-A的诊断;肝脾肿大伴血小板减少、间质性肺病变、血脂异常,尤其是高密度脂蛋白血清水平降低、低密度脂蛋白升高及高甘油三酯血症,提示NPD-B的诊断。
NPD-C 型诊断
一些临床特征可提示NPD-C的诊断,如新生儿期出现的腹水、黄疸消退延迟、肝功能异常、肺间质浸润、持续肌张力过低,幼儿期出现肝脾肿大,后期出现垂直性核上性凝视麻痹、惊厥发作、痴笑猝倒、共济失调、肌张力障碍,成人中可表现为痴呆、抑郁、精神分裂、双相障碍。与 NPD-C强相关临床特征:垂直性核上性凝视麻痹、痴笑猝倒、不明原因的单纯性脾肿大、新生儿黄疸或胆汁淤积时间延长以及过早出现的认知能力下降或痴呆。确诊NPD-C可通过骨髓和皮肤成纤维细胞进行Filipin染色(显示核周集中出现强荧光点,为未酯化胆固醇),另外,外显子测序检出NPD-C1及NPD-C2外显子突变亦可确诊NPD-C型[2]。
动物模型
1.ASM缺乏症(NPD-B)鼠模型
1995年国外有学者,制作了两例ASM基因敲除大鼠模型,纯合子动物模型的网状内皮组织和脑组织中脂质贮积逐渐加重,最主要的就是SPM(神经鞘磷脂),同时还有胆固醇和神经节苷脂。小脑中浦肯野细胞的逐渐减少导致步态异常,6-8月死亡。死亡原因不明,可能继发于神经系统的改变。ASMKO小鼠的肺部感染是主要特点,通过肺部灌洗,证实这些小鼠肺泡中存在大量炎性细胞,并释放炎症因子。对小鼠肺组织的病理检查,发现这些炎症因子可以作为监测疾病进展及治疗效果的指标。
除了完全基因敲除的ASMKO模型外,通过引入SMPD1部分功能基因到ASMKO小鼠身上,一种新的转基因B型NPD小鼠模型也已经产生。这种鼠模型身体大部分器官中ASM活性值为8%,更重要的是,他们没有神经系统症状,具有正常的生存期。然而,这些鼠模型在8-10个月大的时候就开始出现网状内皮系统脂质贮积。这些活体观察数据表明,NPD 患者大脑中低水平的ASM活性有可能起到保护作用,避免神经相关疾病症状的产生,为该病的治疗指明了方向[7]。
2.NPD-C1小鼠模型
6-8周NPD-C1+/−小鼠雌雄配对合笼,交配后获得突变型(NPD-C1-/-)、杂合型(NPD-C1+/-)和野生型(NPD-C1+/+)小鼠[8]。NPD-C1基因突变小鼠(BALB/cNctr-NPD-C1m1N/J,NPD-C1-/-小鼠)的病理改变和临床表现与人的NPD-C1疾病非常相似,NPD-C1突变蛋白缺乏包括固醇感受域在内的C端11个跨膜结构域,从而引起细胞胆固醇转运缺陷,游离胆固醇沉积在溶酶体内。临床表现为中枢神经系统退行性病变,例如震颤、共济失调和发育迟缓等,因此是研究NPD-C1的理想动物模型[9]。

图2 小鼠NPC-1基因型鉴定结果[9]
治疗方式
1.对症治疗
对于NPD-A/B型患者应积极控制肺部感染,缓解呼吸困难,脾功能亢进贫血的患者需补充红细胞。对于NPD-C型患者针对睡眠障碍和惊厥,可以考虑镇静和抗惊厥治疗;针对运动功能减退,给予物理治疗;吞咽困难导致进食困难并因此常引起吸入性肺部感染,可以尝试通过胃肠造瘘术给予胃肠营养[10]。
2.改进的酶替代治疗(ERT)
ERT对非神经性NPD有很好的疗效,可以作为治疗溶酶体病的金标准,但受限于充分糖基化的酶产生的数量及特定部位的识别受体表达的程度,且静脉注射溶酶体酶不能透过血脑屏障。综合各种原因,研究者不得不继续发现新的酶用来治疗NPD,其中一种就是使用细胞粘附分子ICAM1,重组酶至受损部位。使用涂有rASM和抗 ICAM1 抗体的纳米载体进行研究表明,正常细胞和NPD细胞中酶的摄取完全独立于甘露糖-6-磷酸盐受体外,rASM会被迅速转入溶酶体内并在其中有效降解SPM。rASM和抗-ICAM1抗体,目前仍需要进一步考察其免疫原性以及纳米载体本身的生物活性,例如细胞内降解后不产生毒性。在未来的发展中,通过可以识别细胞表面受体的多肽片段可以达到ICAM1治疗的目的——酶的融合和/或单载体的组成[7]。
3.减少底物疗法
葡萄糖苷酰鞘氨醇合成酶(美格鲁特)用于治疗NPD-C型,目前已在欧盟、美国及澳大利亚等国家使用,它能通过血脑屏障,抑制鞘糖脂的合成,降低溶酶体内神经鞘磷脂的沉积,对病变有潜在治疗作用。通过长期的研究和随访证实,美格鲁特可改善或延缓眼球水平跳动、吞咽困难、行走困难等神经系统症状的进展[2]。
4.基因治疗研究
通过在NPD患者体内、外转入SMPD1基因用来表达ASM。基因治疗方法已经广泛应用于ASMKO鼠中。例如,通过逆转录病毒载体,转染SMPD1全长cDNA的至ASMKO骨髓细胞,然后植入局部辐射的ASMKO小鼠体内。网状内皮组织中ASM活性表达水平反映了细胞移植的成功率,从30%到80%不等。
同时,2005年,有学者通过腺病毒载体(AAV)还可以将SMPD1基因植入 ASMKO小鼠的肝细胞中。研究发现ASM活性几近正常,特别在网状内皮系统中,同样的,在大脑中,其无明显作用。作为改进,表达 ASM的AAV 载体直接被注入至中枢神经系统,这样,整个中枢神经系统中ASM活性有了明显提高,进展性共济失调得以控制,生存期恢复正常。上述结果证实,神经性 NPD患者CNS的ASM表达有着很多的作用,但是,必须在神经系统症状出现前注入ASM,否则将无法起效。同时,这种治疗方法的安全性有待进一步考证,也不能确定在其他动物体内ASM活性是否足够。
声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考文献及图片来源:
[1]满永贤.尼曼匹克病误诊1例[J].中国误诊学杂志,2005(02):396-397.
[2]刁倩,黄延风.尼曼-匹克病的研究进展[J].儿科药学杂志,2022,28(07):57-60.DOI:10.13407/j.cnki.jpp.1672-108X.2022.07.015.
[3]https://www.nrdrs.org.cn/app/rare/disease-list-article.html?index=82
[4]https://www.sohu.com/a/60041732_119250
[5]Staatz G,Alibek S,Knerr I,Blessing H,汪玲.尼曼匹克病的影像表现[J].放射学实践,2009,24(06):694.
[6]任守臣,高宝勤.尼曼匹克病C型诊疗新进展[J].中国当代儿科杂志,2015,17(05):533-538.
[7]段佳丽. A型尼曼—匹克病的分子遗传学研究[B].郑州大学,2011.
[8]宋莹,杨记超,李小英,李茜茜,陶子颖,马梦雪,王帅,乔梁,林俊堂.C1型尼曼–匹克病小鼠雄性不育的睾丸病理研究[J].中国细胞生物学学报,2019,41(07):1271-1278.
[9]杨恩慧. C1型尼曼—匹克病小鼠神经系统中髓鞘形成障碍及microRNA差异性表达研究[B].新乡医学院,2017.
[10]https://www.nrdrs.org.cn/app/rare/disease-list-article.html?index=82