

NBAS基因的生物学功能和相关疾病的研究进展
作者:首都医科大学附属北京儿童医院与北京市儿科研究所血液疾病研究室赵宸滋,张晴,李志刚
通信作者:李志刚
文章来源:罕见病研究,2022,1(3):359-364.
【摘要】神经母细胞瘤扩增序列(NBAS)是一个在神经母细胞瘤系发现的高度保守的基因,定位于人类2号染色体p24.3,编码Synaxin-18复合体的一个组成部分,其功能主要是参与高尔基体至内质网的逆向转运和无义介导的mRNA降解。NBAS基因在30余种组织中广泛表达,提示其可能在人体内行使重要的功能。2010年和2015年,NBAS先后被鉴定为SOPH综合征和发热相关急性肝衰竭的致病基因。近期发现,NBAS基因突变也会导致免疫系统受累,引发免疫缺陷。本文就NBAS基因的主要生物学功能、基因突变的致病性及致病机制进行综述。
【关键词】NBAS基因;基因突变;NBAS缺陷病
1999年Wimmer等[1]首次在人神经母细胞瘤细胞系中发现有一个含有CpG岛相关的基因组片段,并将其命名为神经母细胞瘤放大基因(neuroblastoma amplified gene,NAG),该基因常与MYCN(N-myc)基因共扩增。在随后的进一步研究中,该基因又被称之为神经母细胞瘤扩增序列(neuroblastoma-amplified sequence,NBAS)。NBAS基因定位于人类2号染色体p24.3,全长约为420 kb,包含52个外显子,编码一个由2371个氨基酸组成的NBAS蛋白,分子量约为268.57 kDa。NBAS蛋白由一个含有亮氨酸拉链基序的N端结构域(90~371位氨基酸)、一个Sec39结构域(726~1365位氨基酸)和一个功能尚不清晰的C端结构域(1366~2371位氨基酸)组成,该蛋白主要位于内质网、高尔基体和细胞质[2]。NBAS基因及其编码的蛋白在整个身体和发育过程中都有广泛表达[3],提示其可能在人体内行使重要的生物学功能。近期已初步发现NBAS基因的两个主要功能,分别是参与高尔基体至内质网的逆向转运及无义介导的mRNA降解,调节细胞应激反应。
NBAS基因的生物学功能
NBAS基因参与高尔基体至内质网的逆向转运
内质网与高尔基体之间,主要通过囊泡定向运输待转运蛋白。在真核细胞中,新合成的分泌蛋白需要从粗面内质网的间隙定向输送到高尔基复合体进行加工,这一过程称为囊泡由内质网到高尔基体的顺向转运[4]。绝大多数蛋白质将在高尔基体中继续进行其修饰和包装。然而,某些蛋白质需要从高尔基体回收,重新转运至内质网。如在高尔基体上错误折叠的蛋白质等,这一过程称为囊泡的逆向转运[5]。顺向及逆向的膜流动是平衡的,这对维持内质网与高尔基体之间蛋白质和膜磷脂的稳态分布起着至关重要的作用[6]。
上述囊泡的转运过程受到许多因子的调控,其中可溶性N-乙基马来酰亚胺敏感因子结合蛋白受体(soluble ethylmaleimide-sensitive factor attachment pro-tein receptors,SNARE)发挥了重要作用。典型的SNARE蛋白是位于囊泡或靶膜上具有N端胞质结构域的跨膜蛋白,其功能是介导转运囊泡与靶膜的停靠和融合[7]。根据分布位置的不同,SNARE主要分为两类,分别是位于转运囊泡上的v-SNARE(vesicle-membrane SNARE)和位于靶膜上的t-SNARE(target-membrane SNARE),这一对特定的SNARE蛋白之间的特异性识别促进囊泡与靶膜的融合[7]。Syntaxin 18(STX18)是定位于内质网的一个重要t-SNARE,是一种多结构域蛋白,通过与其他t-SNARE蛋白(p31、BNIP1)及外周膜蛋白(ZW10、RINT1、Sly1)共同组成Syntaxin 18复合体[8]。后者是酵母中Ufe1p复合体的同源物,主要功能是介导高尔基体至内质网的囊泡逆向转运[9]。2009年Aoki等[10]研究发现,NBAS基因编码蛋白亦是Syntaxin 18复合体的一个重要亚单位。NBAS蛋白与酵母中Ufe1p复合体的组成亚基Dsl3p/Sec39p具有一定的相似性。在促进SNARE复合物解聚条件下,NBAS可从Syntaxin 18复合体中释放,但仍保留在p31-ZW10-RINT-1亚复合物中。NBAS蛋白与其他成分结合形成Syntaxin 18 复合物,其功能是作为一个栓系蛋白接收从高尔基体逆向转运至内质网的囊泡。研究表明NBAS蛋白的N端和C端区域分别与Syntaxin 18复合体中p31的N端和ZW10-RINT-1相互作用,以桥联p31与ZW10-RINT-1的连接(图1)[10]。敲低NBAS蛋白后,p31的表达水平随之显著下降,证明二者之间的相互作用[10]。2015年,亦有研究在成纤维细胞中观察显示,与对照组相比,NBAS基因突变导致参与内质网应激反应的基因表达显著增加,提示抑制逆行运输可能导致内质网应激反应[11]。然而NBAS在逆向转运中的具体分子机制仍不明确。
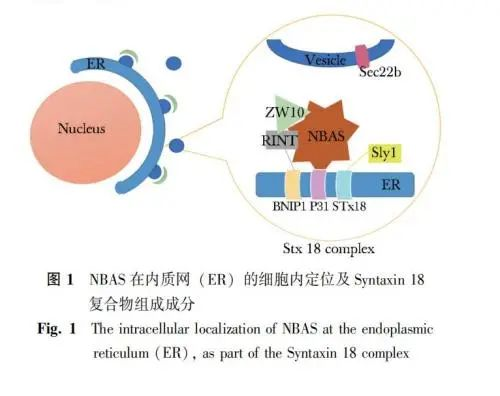
NBAS基因参与无义介导的mRNA降解过程
传统观点认为,无义介导的mRNA降解(nonsense-mediated mRNA decay,NMD)是一种在所有真核生物中高度保守的转录后监测系统,它选择性识别并降解因突变或生物合成错误而产生含有提前终止密码子(premature terminal codon,PTC)的异常mRNAs(无义突变的RNA转录本),其目的是提高基因表达的效率和保真度,可以有效阻止因截短蛋白的产生进而对细胞造成潜在损伤或产生异常的多肽和发挥有害的功能[12]。众多蛋白因子参与NMD过程,目前认为有7个核心NMD因子(UPF1、UPF2、UPF3、SMG1、SMG5、SMG6和SMG7)在降解“异常转录本”和调控“正常转录本”的过程中均发挥了重要作用[13]。
在2007年,Longman等[14]首次通过全基因组RNA干扰扫描技术发现秀丽隐杆线虫体内有一个全新的NMD因子,对应于人的NBAS蛋白。在2011年Anastasaki等[15]研究发现敲除斑马鱼体内的NBAS基因,斑马鱼产生严重的发育缺陷且胚胎存活率显著降低。其表型与核心NMD因子UPF1等缺失引起的严重发育缺陷一致,提示二者可能具有相似的功能,均作为必要蛋白在“异常转录本”的降解过程中发挥关键的作用。随后有研究发现,在斑马鱼中,NBAS直接调节核心NMD因子UPF1的mRNA表达水平,并参与保守的NMD负反馈调控环路,这与哺乳动物细胞中的核心NMD因子的功能保持一致[16-17]。除此之外,在2013年Longman等[18]团队进一步研究发现NBAS在人类、河豚、斑马鱼等中功能高度保守并且在NMD过程中扮演着非常重要的角色。最新研究发现NBAS和UPF1之间存在相互作用,锚定在内质网膜上的NBAS将核心NMD因子UPF1募集到内质网的膜上,从而激活内质网的NMD反应[19]。亦有研究发现NMD除了可以降解“异常转录本”,还能够调节细胞中10%~20%具有NMD诱导功能的天然mRNAs表达,即也对维持正常基因表达调控的动态平衡、细胞生长和发育具有重要的意义[20-21]。不仅如此,Longman等[18]在Hela细胞系中发现NBAS可以调控多达1444个基因的表达,约占所有表达基因总数量的10%,其中590个基因表达上调,854个基因表达下调,提示NBAS基因对调控“正常转录本”表达也发挥作用。尽管NBAS在NMD过程中发挥重要作用,但具体的机制尚不清楚,仍需要进一步研究。
NBAS基因缺陷相关疾病
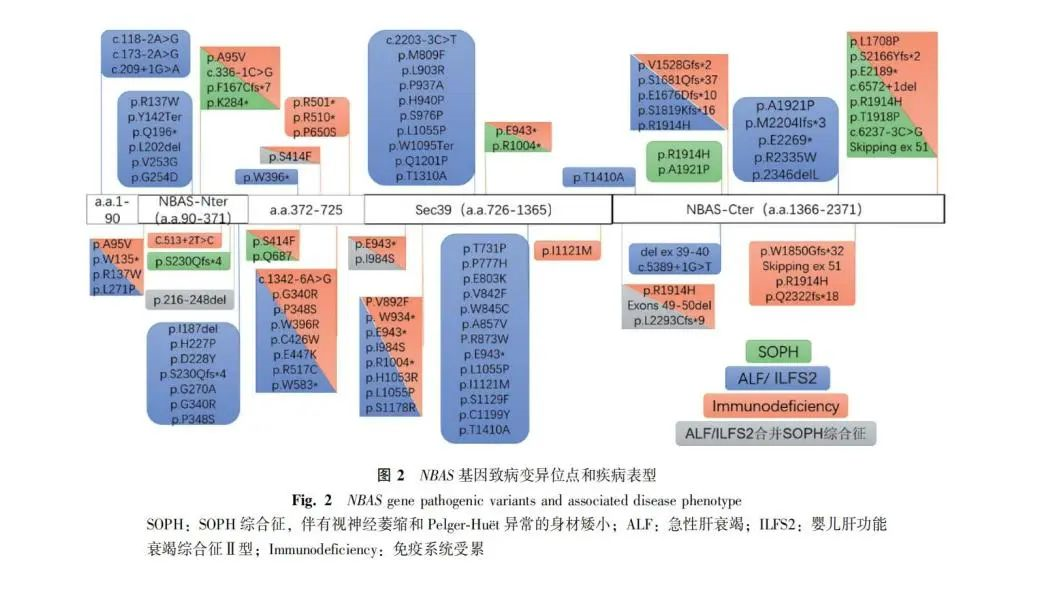
近年来,许多文献报告了NBAS基因突变可导致一些罕见的常染色体隐性遗传病,但临床表现具有较大的异质性,可表现为孤立或累及多器官系统的混合型临床特征,以SOPH综合征、发热相关急性肝衰竭(acute liver failure, ALF)常见[22]。2010年Maksimova等[23]首次报道了NBAS基因缺陷可导致身材矮小、视神经萎缩和Pelger-Hut畸形综合征,主要表现为严重的生长发育迟缓、身材矮小、老年样面部畸形、Pelger-Huët异常、视神经萎缩,并根据其临床特征统称为SOPH综合征。该团队通过全外显子测序技术鉴定出NBAS基因C端结构域的p.Arg1914His(c.5741G>A)位点纯合错义突变。2015年Haack等[11]首次通过全外显子测序技术先后在4例反复急性肝衰竭(recurrent acute liver failure, RALF)和11例ALF患儿中发现了不同位点的NBAS纯合或复合杂合突变,提示ALF与罕见的NBAS基因缺陷有关。值得注意的是,NBAS缺陷患者在RALF或ALF前常伴有发热,随后出现肝脏转氨酶水平持续升高、黄疸及肝功能障碍,提示发热可能是导致这些NBAS突变患者发生RALF或ALF的原因[24]。目前,至少已有来自112个家系的126例NBAS缺陷病患者在全球范围内被报道,涉及101种不同的NBAS变异[25],其中位于NBAS蛋白N端结构域的c.5741G>A(p.Arg1914His)错义突变是最常见的NBAS变异类型,多见于雅库特人群。值得注意的是,迄今未见有携带双等位基因纯合或复合杂合无义突变的患者,提示完全丧失NBAS基因功能可能是致命的。2019年Carli和 Staufner团队先后回顾分析了73例及110例NBAS基因缺陷患者的临床特征,并探究其基因型与临床表型的关系[22,26]。上述患者主要表现为三种临床亚型,并可能与NBAS基因变异位置直接相关:SOPH综合征的突变位点普遍位于NBAS蛋白C端结构域;ALF和婴儿肝功能衰竭综合征Ⅱ型(infantile liver failure syndrome type 2, ILFS2)的突变位点集中在NBAS蛋白的Sec39结构域及N端结构域;而NBAS蛋白N端β-螺旋体结构域变异的患者则可以呈现一种ALF/ILFS2合并SOPH综合征的多系统混合表型。2020年,Li等[27]亦回顾分析了国内24例NBAS缺陷病患者的临床和基因特征。与前期研究结果相似,该发现携带变异位于NBAS蛋白Sec39结构域的患者更易发生ALF(100.0% vs. 12.5%,P=0.000),而携带变异位于NBAS蛋白C端的患者主要表现为肝外多系统的受累(40% vs. 100%,P=0.0128)。并且,c.3596G>A/p.Cys1199Tyr错义突变可能是ALF患者的热点突变,在国内24例患者中9例均表现为此突变。上述研究对指导患者的临床治疗及预测预后具有重要的意义。然而,上述基因型与表型之间的相关性并非绝对的,除了基因变异位置,NBAS基因的变异性质、其他遗传或环境因素的累积可能也影响了患者临床表型。根据已报道的NBAS基因致病变异位点和疾病表型,本文对大部分基因突变位点进行梳理总结(图2)。
值得注意的是,NBAS基因突变也会导致免疫系统受累。据统计,约60%的NBAS缺陷患者同时伴有免疫症学症状或实验室检测异常,如反复感染、低丙种球蛋白血症、自然杀伤细胞数量减少等[23]。其突变位点主要位于NBAS蛋白N端和C端结构域[26]。然而,针对NBAS基因缺陷病的特异性免疫表型或免疫功能的改变,目前尚无全面的系统性分析。2015年,Segarra等[28]报道了两例因反复病毒和细菌感染而导致肺炎和中耳炎的患者,其在接种疫苗后机体出现了低丙种球蛋白血症、自然杀伤细胞减少和抗体产生异常的免疫反应。该报道首次发现NBAS基因突变与机体免疫缺陷相关。2019年,Ricci等[29]报告在新生儿免疫缺陷病常规筛查中发现一例携带NBAS基因缺陷患儿,其临床主要以联合免疫缺陷为主,未发现ALF和SOPH综合征相关表型,提示NBAS基因变异可导致先天性免疫缺陷病。近期,Lenz等[30]深入分析了15例确诊NBAS基因缺陷病患儿的免疫学检测指标,结果显示所有患儿均具有相似的免疫异常特征,即外周血naïve B细胞数量和自然杀伤细胞绝对数显著减少,且自然杀伤细胞毒脱颗粒功能及对靶细胞的杀伤能力明显降低。随后,Albar等[31]亦报告一例NBAS基因突变相关自然杀伤细胞缺乏症患儿,临床表现为反复的病毒及细菌多发感染,在定期输注干扰素β-1a后病情好转,但停药后复发。以上研究进一步提示NBAS可能是一种先天免疫缺陷病的致病基因。近期,国际免疫学联合会(The International Union of Immunological Scienties,IUIS)在先天性免疫缺陷病分类中亦关注到NBAS基因缺陷。
NBAS基因变异所致疾病虽然复杂,但其诊断主要依赖于患者的临床表现及基因检测。如发现患儿具有SOPH综合征临床表型、不明原因的反复肝酶异常或反复ALF、免疫异常等症状时,需警惕此病,及时进行基因检测,防止漏诊和误诊。对于NBAS基因变异所致疾病相对罕见,治疗暂无相关文献报道,应根据患者的病情进行个体化治疗,而患者的预后及其生存质量尚不清楚,应定期随访和进一步研究。
NBAS基因缺陷病的致病机制
目前NBAS基因变异相关疾病仍以临床散发病例报告为主,确切发病机制尚不明确,有待进一步研究。有文献称,NBAS介导的NMD调节功能与骨骼发育和胆固醇生物合成存在紧密联系,这可能是SOPH综合征患儿临床表现出骨骼发育不良和Pelger-Huet异常的重要原因[15]。研究发现,在携带NBAS突变的ALF患者的皮肤成纤维细胞中,NBAS蛋白及Syntaxin 18复合体的组成亚基p31蛋白的表达显著降低,顺式高尔基体及内质网-高尔基体中间隔室结构减少,同时调控内质网应激反应的相关基因表达显著增加。据此推测NBAS突变可能导致高尔基体至内质网的逆向转运受阻,诱发内质网应激反应,进而引起肝细胞凋亡及肝功能衰竭[11,32]。由于NBAS缺陷相关ALF常以发热性疾病诱发ALF为特征,研究发现NBAS突变蛋白具有热敏感性,高温条件下其稳定性显著下降。因此,推测发热期间高能耗需求的分解代谢状态及温度升高本身可能进一步使NBAS蛋白表达减少,内质网和高尔基体之间的囊泡运输环节受到干扰,进而加剧内质网应激反应诱发ALF[11,33]。此外,亦有研究报道携带RINT1基因突变患者也具有类似ALF的临床表型。RINT1突变细胞伴有自噬小体成熟障碍,自噬潮受抑制,因此,推测肝脏中的自噬受损可能导致肝脏脂肪变性、肝细胞纤维化,最终引发肝脏疾病[34]。由于NBAS蛋白C端可与Syntaxin 18复合体中的ZW10-RINT-1亚单位相互作用,因此,NBAS缺陷相关ALF的发生机制可能也与自噬改变相关。
结语
总之,NBAS基因参与机体一些重要的生理过程,对于维持内质网的动态平衡及无义RNA的清除具有重要意义。NBAS基因缺陷病临床表型复杂多样,相关发病机制还未完全阐明,还需要进一步在分子生物学层面的研究,以期探索潜在的治疗靶点。
参考文献:
[1]Wimmer K, Zhu XX, Lamb BJ, et al. Co-amplification of a novel gene, NAG, with the N-myc gene in neuroblastoma[J]. Oncogene, 1999, 18: 233-238.
[2]Scott DK, Board JR, Lu X, et al. The neuroblastoma amplified gene, NAG: genomic structure and characterisation of the 7.3 kb transcript predominantly expressed in neuroblastoma[J]. Gene, 2003, 307: 1-11.
[3]Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome[J]. Science, 2015, 347: 1260419.
[4]Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion[J]. Cell, 2004, 116: 153-166.
[5]Sannerud R, Saraste J, Goud B. Retrograde traffic in the biosynthetic-secretory route: pathways and machinery[J]. Curr Opin Cell Biol, 2003, 15: 438-445.
[6]Spang A. Retrograde traffic from the Golgi to the endoplasmic reticulum[J]. Cold Spring Harb Perspect Biol, 2013, 5: a013391.
[7]Hong WJ, Lev S. Tethering the assembly of SNARE complexes[J]. Trends Cell Biol, 2014, 24: 35-43.
[8]Hirose H, Arasaki K, Dohmae N, et al. Implication of ZW10 in membrane trafficking between the endoplasmic reticulum and Golgi[J]. EMBO J, 2004, 23: 1267-1278.
[9]Lewis MJ, Pelham HR. SNARE-mediated retrograde traffic from the Golgi complex to the endoplasmic reticulum[J]. Cell, 1996, 85: 205-215.
[10]Aoki T, Ichimura S, Itoh A, et al. Identification of the neuroblastoma-amplified gene product as a component of the syntaxin 18 complex implicated in Golgi-to-endoplasmic reticulum retrograde transport[J]. Mol Biol Cell, 2009, 20: 2639-2649.
[11]Haack TB, Staufner C, Köpke MG, et al. Biallelic mutations in NBAS cause recurrent acute liver failure with onset in infancy[J]. Am J Hum Genet, 2015, 97: 163-169.
[12]Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination[J]. Nat Rev Mol Cell Biol, 2012, 13: 700-712.
[13]Hug N, Longman D, Cáceres JF. Mechanism and regulation of the nonsense-mediated decay pathway[J]. Nucleic Acids Res, 2016, 44: 1483-1495.
[14]Longman D, Plasterk RHA, Johnstone IL, et al. Mechanistic insights and identification of two novel factors in the C. elegans NMD pathway[J]. Genes Dev, 2007, 21: 1075-1085.
[15]Anastasaki C, Longman D, Capper A, et al. Dhx34 and Nbas function in the NMD pathway and are required for embryonic development in zebrafish[J]. Nucleic Acids Res, 2011, 39: 3686-3694.
[16]Huang L, Lou CH, Chan W, et al. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD[J]. Mol Cell, 2011, 43: 950-961.
[17]Yepiskoposyan H, Aeschimann F, Nilsson D, et al. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells[J]. RNA, 2011, 17: 2108-2118.
[18]Longman D, Hug N, Keith M, et al. DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans[J]. Nucleic Acids Res, 2013, 41: 8319-8331.
[19]Longman D, Jackson-Jones KA, Maslon MM, et al. Identification of a localized nonsense-mediated decay pathway at the endoplasmic reticulum[J]. Genes Dev, 2020, 34: 1075-1088.
[20]Nicholson P, Yepiskoposyan H, Metze S, et al. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors[J]. Cell Mol Life Sci, 2010, 67: 677-700.
[21]Palacios IM. Nonsense-mediated mRNA decay: from mechanistic insights to impacts on human health[J]. Brief Funct Genomics, 2013, 12: 25-36.
[22]Staufner C, Peters B, Wagner M, et al. Defining clinical subgroups and genotype-phenotype correlations in NBAS-associated disease across 110 patients[J]. Genet Med, 2020, 22: 610-621.
[23]Maksimova N, Hara K, Nikolaeva I, et al. Neuroblastoma amplified sequence gene is associated with a novel short stature syndrome characterised by optic nerve atrophy and Pelger-Hut anomaly[J]. J Med Genet, 2010, 47: 538-548.
[24]Li JQ, Qiu YL, Gong JY, et al. Novel NBAS mutations and fever-related recurrent acute liver failure in Chinese children: a retrospective study[J]. BMC Gastroenterol, 2017, 17: 1-7.
[25]Ritelli M, Palagano E, Cinquina V, et al. Genome-first approach for the characterization of a complex phenotype with combined NBAS and CUL4B deficiency[J]. Bone, 2020,140:115571.
[26]Carli D, Giorgio E, Pantaleoni F, et al. NBAS pathogenic variants: defining the associated clinical and facial phenotype and genotype-phenotype correlations[J]. Hum Mutat, 2019,40:721-728.
[27]Li ZD, Abuduxikuer K, Zhang J, et al. NBAS disease: 14 new patients, a recurrent mutation, and genotype-phenotype correlation among 24 Chinese patients[J]. Hepatol Res, 2020, 50: 1306-1315.
[28]Segarra NG, Ballhausen D, Crawford H, et al. NBAS mutations cause a multisystem disorder involving bone, connective tissue, liver, immune system, and retina[J]. Am J Med Genet A, 2015, 167: 2902-2912.
[29]Ricci S, Lodi L, Serranti D, et al. Immunological features of neuroblastoma amplified sequence deficiency: report of the first case identified through newborn screening for primary immunodeficiency and review of the literature[J]. Front Immunol, 2019, 10: 1955.
[30]Lenz D, Pahl J, Hauck F, et al. NBAS Variants Are Associated with quantitative and qualitative NK and B cell deficiency[J]. J Clin Immunol, 2021, 41: 1781-1793.
[31]Albar RF, Alsulimani EF, Alsalmi KA, et al. Natural killer cell deficiency in neuroblastoma amplified sequence gene mutation[J]. Cureus, 2021, 13:e19270.
[32]Staufner C, Haack TB, Köpke MG, et al. Recurrent acute liver failure due to NBAS deficiency: phenotypic spectrum, disease mechanisms, and therapeutic concepts[J]. J Inherit Metab Dis, 2016,39:3-16.
[33]Cotrina-Vinagre FJ, Rodríguez-García ME, Martín-Hernández E, et al. Characterization of a complex phenotype (fever-dependent recurrent acute liver failure and osteogenesis imperfecta) due to NBAS and P4HB variants[J]. Mol Genet Metab, 2021,13:201-210.
[34]Cousin MA, Conboy E, Wang JS, et al. RINT1 Bi-allelic variations cause infantile-onset recurrent acute liver failure and skeletal abnormalities[J]. Am J Hum Genet, 2019, 105: 108-121.
文章来源于罕见病研究编辑部