

Science|人类和灵长类动物中可耐受的遗传变异图谱
发布时间: 2023-06-08
介绍:
到目前为止,数百万人已经接受了全基因组和全外显子组测序,这是一个巨大的投入,首次揭示了在物种内作为个体区分遗传差异的广泛目录。然而,大多数这些基因变异的影响仍然未知,限制了它们的临床实用性和可操作性。能够准确区分致病突变和良性突变,并在全基因组范围内解释基因变异的新方法,将构成实现个性化基因组医学潜力的有意义的初始步骤。

图1.science官网截图(图片来源于基因检测与解读)
由于人类和非人类灵长类动物之间的进化距离短,该研究的蛋白质几乎完美地分享了相似的氨基酸序列。因此,一旦在同一物种中发现蛋白质突变的影响,这种影响很可能在另一物种中也存在。通过系统地记录非人类灵长类动物的常见变异,研究旨在将这些变异注释为不太可能引起人类疾病的变异,因为在密切相关的物种中这些变异已被自然选择所容忍。一旦数据被收集,该资源的结果可用于使用机器学习来推断全基因组中未观察到的变异的影响。
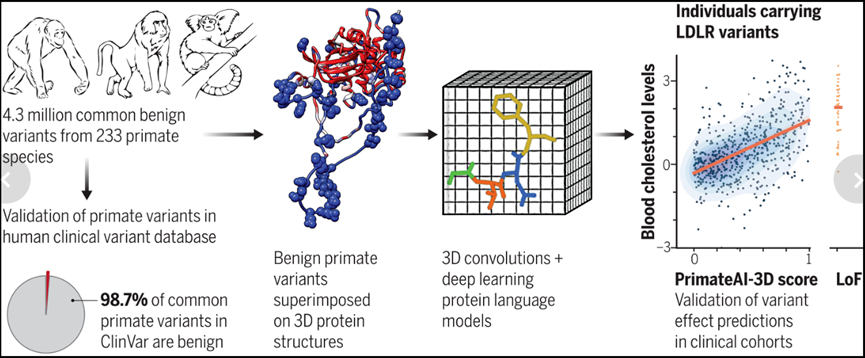
图2.来源于基因检测与解读
结果:
为了推断人类基因组中剩余错义突变的致病性,研究开发了一个半监督三维卷积神经网络,名为PrimateAI-3D,可运行于体素化的蛋白质结构上。使用半监督学习将PrimateAI-3D训练为能够在三维空间中分离常见的灵长类动物变异和相应的对照变异。将经过训练的PrimateAI-3D模型与15个其他已发表的机器学习方法一起评估,以评估它们在六个不同的临床基准测试中区分良性和致病变异的能力。结果表明,PrimateAI-3D在每个任务中表现均优于所有其他算法。
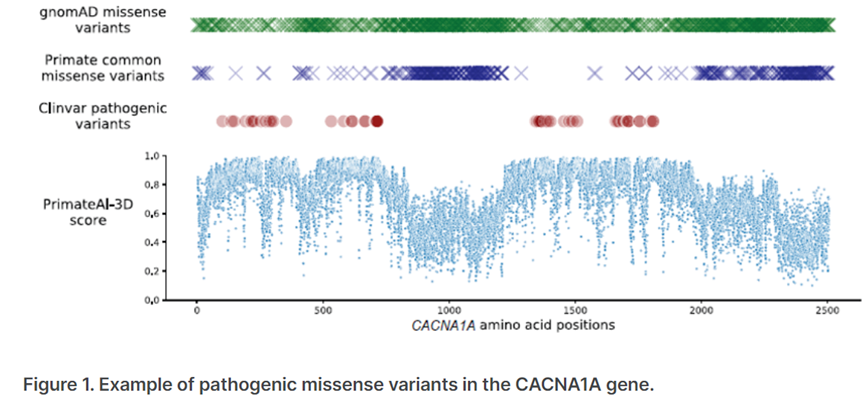
图3.来源于基因检测与解读
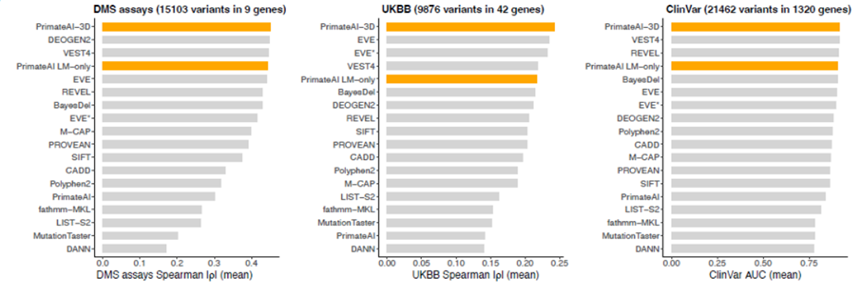
图4.来源于基因检测与解读
结论:
文章来源于基因检测与解读