

Nature | 在“100000基因组计划”中发现罕见病基因关联
一、研究概述
2025年2月26日,伦敦玛丽女王大学(Queen Mary University of London)的Damian Smedley教授在Nature期刊发表标题为“Rare disease gene association discovery in the 100,000 Genomes Project”的研究论文。该研究开发了一种针对孟德尔疾病罕见变异的基因负担分析框架,并将其应用于“10万基因组计划”中34,851个病例及其家庭成员的全基因组测序数据,发现了141个新的疾病-基因关联。

罕见病在全球范围内影响3.5%到5.9%的人口。尽管基因组测序技术取得了进展,仍有50%到80%的患者在遗传门诊中无法获得分子诊断。此外,在《人类孟德尔遗传在线》(OMIM)数据库中记录的10,000种罕见孟德尔病中,不到一半的疾病已明确其遗传基础。诊断失败可能是因为缺乏对非编码区或结构变异的常规筛查。然而,很可能许多未确诊的罕见病患者(病例)的致病变异存在于尚未被发现的基因中,这些基因可能与(非常罕见的)疾病相关。
罕见病测序研究的规模,例如“未确诊疾病网络”、“孟德尔基因组中心”、“发育障碍解读”和“10万基因组计划”(100KGP),为深入了解遗传病的致病机制提供了更多机会,包括通过病例-对照分析建立疾病-基因关联的可能性,这种方法之前曾被用于识别影响复杂疾病风险的常见遗传变异。这种方法为识别携带罕见致病变异的基因提供了急需的统计能力。
为了识别与疾病相关的基因,作者最近开发了一个框架,利用Exomiser变异优先级工具识别的罕见蛋白编码变异,在100KGP数据的初步版本中,对单个先证者(家族中首个被识别为患有罕见遗传病的人)及其家庭成员相对于对照家庭进行基于基因的负担测试。在此前的研究中,通过计算机辅助筛选,作者突出了22个新的疾病-基因关联,其中3个也在独立研究中被报道。
在本研究中,作者进一步优化了基因负担分析框架,改进了罕见变异过滤和统计建模,使其更适合孟德尔疾病和罕见事件的不平衡病例-对照研究,并将其扩展为适用于任何大规模罕见病测序队列的通用工具,补充了可视化脚本,并将其作为开源R分析框架geneBurdenRD发布。此外,作者报告了该方法在100KGP最终数据的更大队列中的应用,包括34,851个家庭、226种罕见病和4,643,230个罕见候选变异的起始库,并通过改进的计算机辅助筛选和增加临床专家筛选,识别出69个可能的新疾病-基因关联。
二、正文
1. 基因负担分析框架
作者开发了一个开源的R框架,用于对罕见病测序队列中的用户定义病例与对照组进行基因负担测试。
该框架的输入包括:(1)罕见致病变异文件,(2)病例-对照分析标签文件,以及(3)样本标识符和病例-对照分配文件。病例和对照可通过疾病类别、表型注释或表型聚类定义。
框架通过Firth逻辑回归模型评估假发现率(FDR)校正后的疾病-基因关联,测试的变异类型包括:(1)预测的失功能(LoF)变异;(2)高度预测的致病性变异;(3)约束性编码区域(CCR)中的变异;以及(4)新生变异。输出结果包括火山图、棒棒糖图和HPO注释的分层分布图,用于可视化分析结果。
该框架适用于大规模罕见病测序队列分析,支持本地或HPC集群运行。用户需准备输入文件并根据疾病数量调整脚本。更多细节和示例数据可在GitHub页面查看。
2. 100KGP的应用
本研究对100KGP罕见病项目中的34,851个单个先证者和大家庭(共72,690个基因组)进行了罕见变异基因负担分析(图1)。该队列的性别和遗传推断祖先分布与英国人口的种族分布基本一致。通过Exomiser分析和变异质量控制,生成了包含4,643,230个罕见、蛋白编码、分离性且高致病性预测变异的起始库。研究对226种“特定疾病”的病例与20个宽泛“疾病组”的对照进行分析,检测了LoF、高致病性、CCR区域的高致病性以及新生变异等类别中基因的显著富集。共进行了161次病例-对照基因负担分析,仅考虑病例中频率高于对照的基因富集。
在0.5%假发现率(FDR)的阈值下,研究识别出165个已知和141个新的潜在疾病-基因关联。其中,5个新关联在初步评估后获得了独立支持证据。其余潜在关联经过进一步筛选,最终确定了69个关联(图1)。这些关联的变异根据ACMG标准自动分类。结合文献综述和表型证据,研究识别了30个有实验支持的关联,其中27个的致病性证据为中等,其余为有限(图1)。
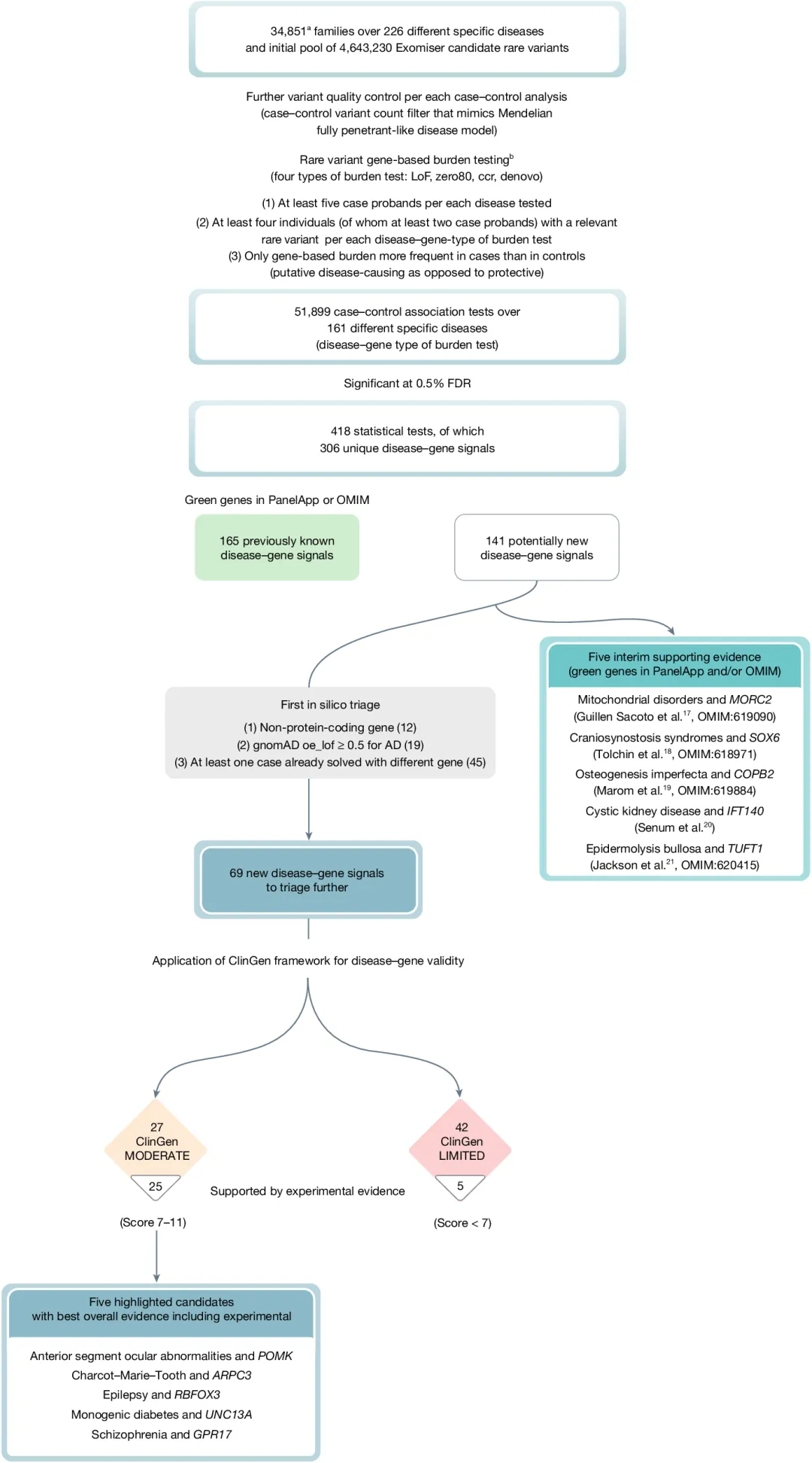
图1. 100KGP数据的稀有变异基因负荷分析。
3. 单基因糖尿病与UNC13A的关联研究
研究发现UNC13A基因变异与“具有单基因病因提示的糖尿病附加表型”之间存在显性关联(ClinGen评分为9,中等)。这一关联由两个单例病例中的罕见预测失功能(LoF)变异驱动,这两种变异均未出现在gnomAD v.4.1.0数据库中,被分类为“可能致病性”,并预测会通过无义介导的衰变(NMD)机制降解(调整后P值为0.0005,比值比为329.8;图2a)。
UNC13A基因在gnomAD中罕见LoF变异耗竭(观察/预期LoF比值为0.09,LoF不耐受概率pLI为1)。研究表明,UNC13A在调节β细胞功能中发挥重要作用。从UNC13A基因敲除小鼠中提取的新生胰腺β细胞表现出对葡萄糖刺激的胰岛素分泌受损,杂合小鼠敲除模型表现出葡萄糖耐受不良。此外,UNC13A与已知单基因糖尿病基因的共表达网络分析显示,在胰腺组织中显著富集(FDR校正后的P值为0.01)。
然而,预测的LoF变异也在无糖尿病病史的对照组中被观察到,表明这些变异的外显率不完全、发病时间较晚,或者对照组中的变异并非真正的LoF变异。
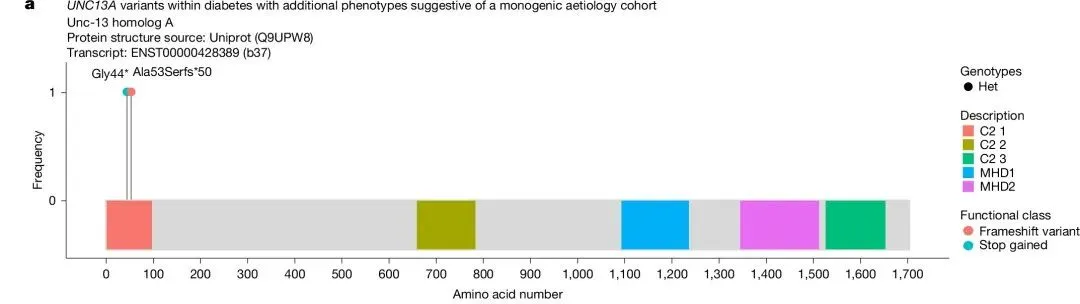
图2. 疾病-基因关联的证据。
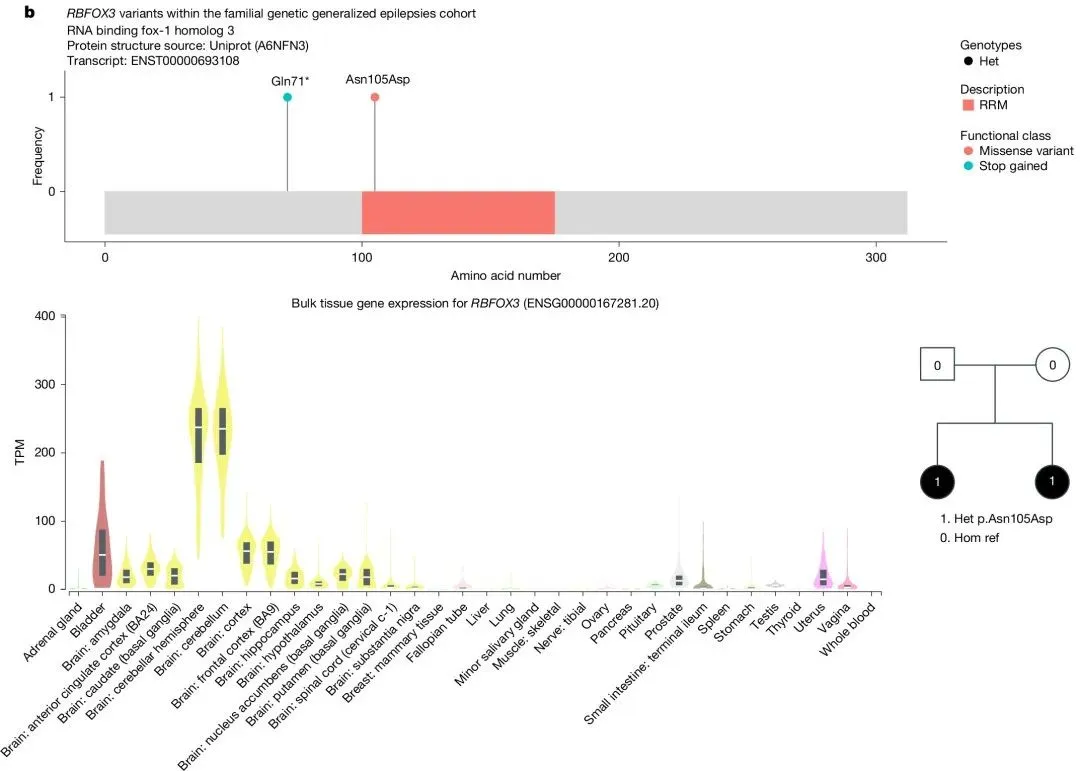
4. 癫痫与RBFOX3的关联
研究发现RBFOX3基因变异与“家族性遗传性全面性癫痫”之间存在显性关联(ClinGen评分为11,中等)。这一关联由两个罕见的预测致病变异驱动:两个受影响姐妹中的p.Asn105Asp变异和一个伴有学习障碍表型的先证者中的p.Gln71*变异(图2b)。这两种变异均未出现在gnomAD v.4.1.0和ClinVar数据库中,被分类为意义不明的变异(VUS)。
RBFOX3在大脑中特异性表达,尤其是在小脑中。小鼠模型显示,RBFOX3变异会增加癫痫发作的易感性。尽管RBFOX3与癫痫之间的潜在关联早在2013年就已发表,但OMIM或PanelApp尚未收录该关联,且基因策展联盟(GenCC)记录的证据有限。本研究进一步支持了RBFOX3与癫痫的关联,并结合近期研究结果,表明RBFOX3在癫痫调控中发挥关键作用,可能成为一种潜在的治疗靶点。
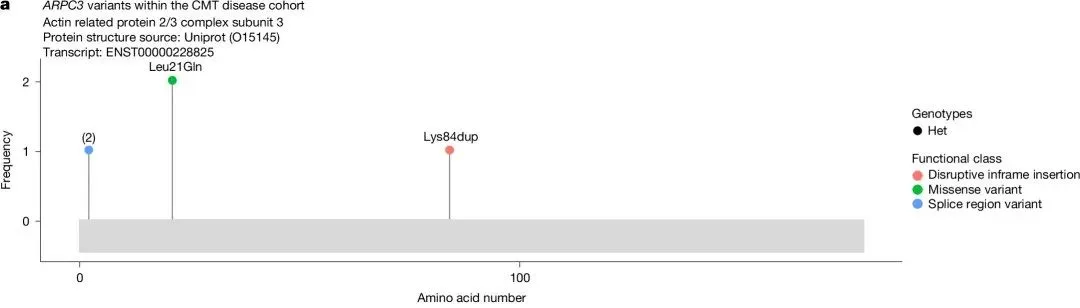
5. Charcot–Marie–Tooth病与ARPC3的关联
研究发现ARPC3基因变异与Charcot–Marie–Tooth(CMT)病之间存在关联(ClinGen评分为8,中等)。这一关联由四个病例中的罕见杂合变异驱动,包括p.Leu21Gln、p.Lys84dup和c.6G>C变异(3a)。这些变异在gnomAD数据库中极为罕见或缺失。
这四个病例表现出高度的表型相似性(PhenoDigm平均得分为0.84),其特征为上下肢远端肌肉无力和周围轴突神经病变。在1000个随机抽样的相同大小的CMT病例集中,仅有10个达到相同的平均得分或更高,表明ARPC3相关家族在表型上与其他CMT病例显著不同。
ARPC3与已知CMT基因DNM2和SYT2存在蛋白-蛋白相互作用。微管在神经细胞骨架中发挥关键作用,其失调与多种神经系统疾病相关,ARPC3可能通过miR-29a/b下游调节树突棘形态。条件性敲除ArpC3的小鼠表现出轴突包裹失败。
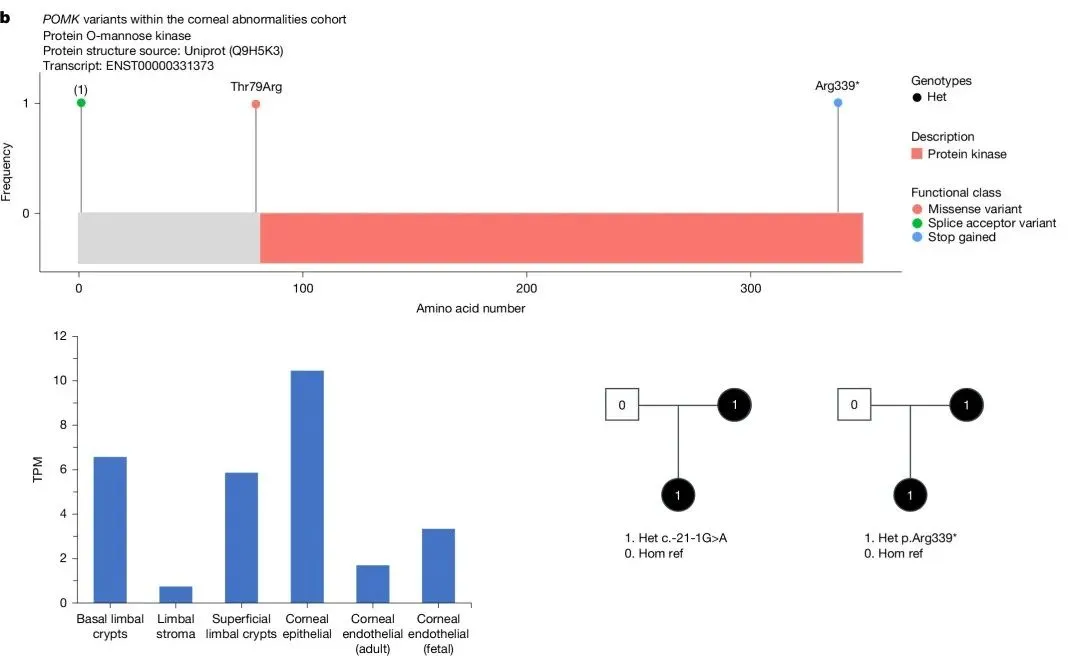
6. 角膜异常与POMK的关联
研究发现POMK基因变异与“角膜异常”之间存在显性关联(ClinGen评分为9,中等)。这一关联由三个病例中的罕见预测致病变异驱动,包括两个预测的LoF变异和一个错义变异,其表型提示为前段发育不良(ASD;图3b)。ASD是一组影响眼前节的发育障碍,通常具有不完全外显率和表现度变异。
在两个三口之家的病例中观察到共分离现象,其中女性先证者从受影响的母亲那里遗传了杂合的剪接受体变异c.-21-1G>A(gnomAD v.4.1等位基因频率为0.000011)和杂合的移码终止变异p.Arg339*(gnomAD v.4.1等位基因频率为0.000001)。这两种预测的LoF变异均被分类为VUS(意义不明的变异)。此外,在一个单例病例中观察到一个杂合的错义变异p.Thr79Arg(gnomAD v.4.1等位基因频率为0.000011),并被分类为“可能良性”。
POMK参与α-肌聚糖的O-连接糖链的呈现,该糖链与细胞外基质和细胞骨架之间形成跨膜连接。RNA-seq数据表明,POMK在角膜上皮中表达量最高(图3b)。POMK的双等位LoF变异与常染色体隐性肌营养不良性糖蛋白病相关,这种疾病包括多种眼部异常,表明POMK在眼部发育中发挥关键作用。斑马鱼中敲低pomk基因的实验也显示出发育性眼部异常。本研究中识别的变异是否通过POMK单倍剂量不足或显性功能获得效应诱导ASD,值得进一步研究。
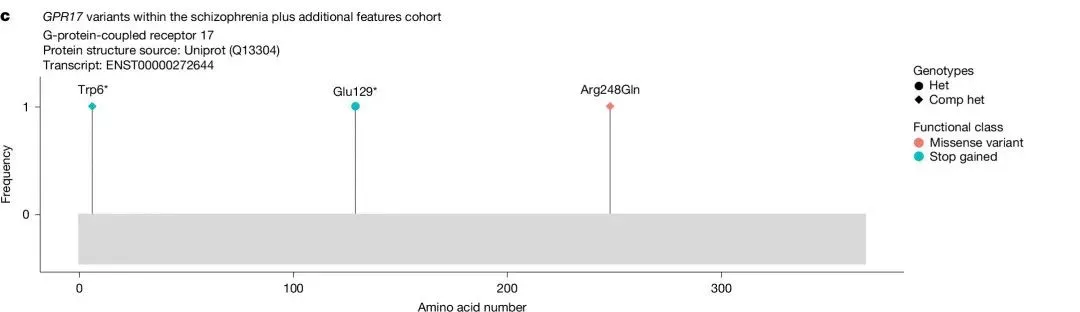
7. 精神分裂症与GPR17的关联
研究发现GPR17基因变异与“精神分裂症附加特征”之间存在关联(ClinGen评分为9,中等)。这一关联由两个单例病例中的罕见预测失功能(LoF)变异驱动,这两个病例均表现出精神分裂症和其他精神症状。具体变异包括:第一个病例为p.Trp6*变异(gnomAD v.4.1等位基因频率为0.00018),与p.Arg248Gln变异呈复合杂合状态;第二个病例为杂合的p.Glu129*变异(gnomAD v.4.1等位基因频率为0.00001)。这两种变异均预测不会发生无义介导的衰变(NMD;图3c)。
gnomAD数据显示(LoF观察/预期值为0.79 [0.45–1.46],pLI值为0),不支持该基因的单倍剂量不足机制。这可能是因为变异以隐性方式作用,或者高LoF观察/预期值可能反映了疾病的晚发性、不完全外显率特征和/或多基因机制。GPR17在大脑中表现出高度特异性表达,尤其是在大脑皮层。研究表明,GPR17能够调节少突胶质细胞的分化和髓鞘化,这一过程在包括精神分裂症在内的多种神经系统疾病中发挥重要作用。
来源:BioMedDaily
【声明】本文为转载文章,本平台仅作分享、传递信息,版权归原作者所有,如有侵权,请联系删除。