

RDDC邀您关注Leber先天性黑矇:瞳光之殇
疾病概述
Leber先天性黑矇(Leber Congenital Amaurosis, LCA)是一种极其罕见的遗传性视网膜疾病,以其严重的致盲性特征而著称。患者往往在年幼时便出现症状,视功能受到显著损害。台湾学者的一项研究揭示了LCA患者的发病年龄普遍偏早,并且他们常在青少年时期寻求医疗帮助[1]。早在1869年,Theodore Leber博士便首次描述了患有眼球震颤和瞳孔光反射异常的婴儿所展现出的严重视力问题,这被认为是LCA的典型表征[2]。
此外,流行病学报道显示LCA占据了全部遗传性视网膜营养不良症的5%,其患病率约为每81,000至30,000人中就有1人受到影响。此外,LCA还占据了学龄儿童失明原因的20%,显示了其对儿童视力的严重威胁[1,3-5]。
表型特点与诊断标准
LCA的主要特征显著,包括:在出生或出生后不久即展现出的严重视力丧失,常伴随眼球震颤(不自主的眼球运动)、瞳孔反射迟钝、畏光或夜盲等症状。其视网膜电图(ERG)显示,各波形记录几乎不存在或显著降低。LCA通常在患儿出生后6个月内发病,家长往往因发现孩子眼球震颤、无法注视或斜视而寻求医疗帮助[6]。
除了上述症状外,LCA还有其他一些常见的临床表现。如图1所示,患者的眼底损伤表现为视网膜血管组织减少,视网膜变薄,黄斑区出现变化,且缺乏中央暗斑。此外,患者还可能面临视力持续下降、屈光不正等问题[7]。这些特征为医生提供了诊断和治疗LCA的重要依据。
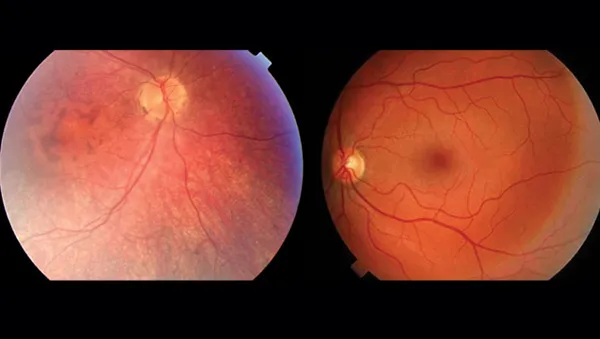
图1. LCA患者与健康人眼底表现比较(左为LCA患者,右为正常人)
来源:澎湃新闻
关于LCA诊断标准[8],具体如下:
(1)患者6月龄前出现严重视力低下或盲,可伴有眼球震颤、指眼征、黑瞳孔等;
(2)ERG各波形记录不到或严重降低;
(3)不伴有或伴有其他眼部或其他系统的先天发育异常。
致病基因
根据临床验证和遗传学分析,共有23个基因与LCA相关,其中最常见的基因为GUCY2D、RPE65、CRB1、CEP290等[9]。
⭐️ GUCY2D
GUCY2D(图2)是第一个被鉴定为与LCA相关的基因,位于染色体17p13.1上,编码视网膜鸟苷酸环化酶-1,该酶参与光转导中的感光器恢复阶段。GUCY2D突变占LCA病例的6-21%。已知GUCY2D相关LCA病例在生命早期视力极差但静止不动,伴有眼球震颤、眼指征和明显的畏光[10,11]。
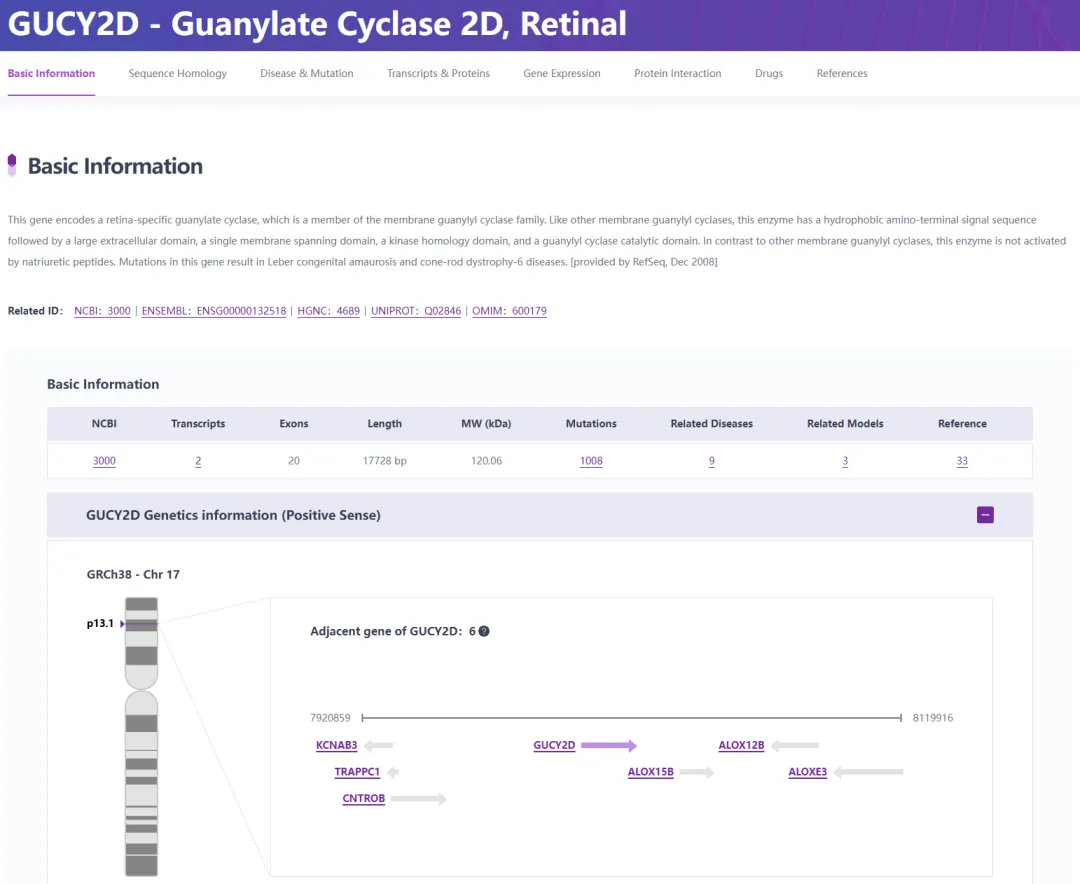
图2. GUCY2D基因概况
来源:RDDC罕见病数据中心
⭐️ RPE65
染色体1p31.3上的RPE65基因编码类视黄醇异构酶(图3),这是一种在RPE中大量表达的61kDa酶,负责类维生素A循环中的维生素A代谢。这种缺陷导致缺乏11-顺式视网膜再生,从而影响视力。RPE65突变占LCA病例的4%-16%,在中国人群中相对罕见,但在高加索和印度人群中更为普遍[12]。
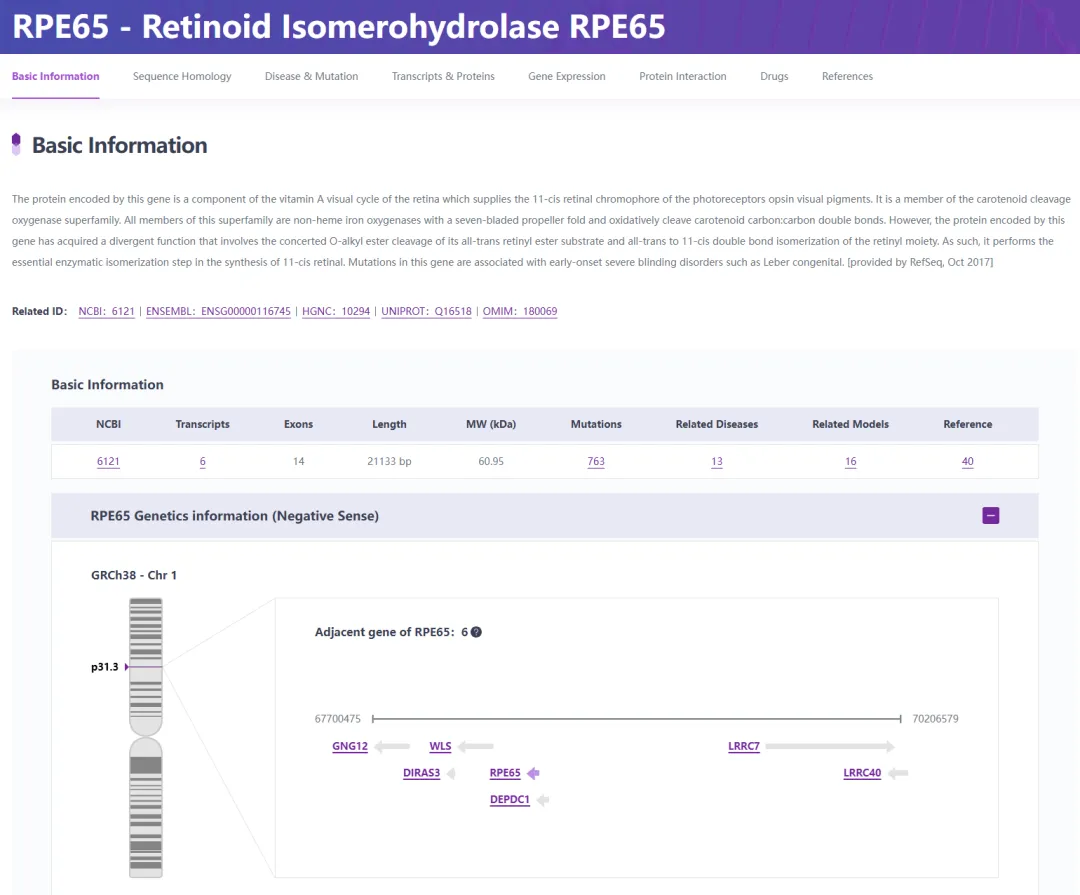
图3. RPE65基因概况
来源:RDDC罕见病数据中心
该基因与LCA2最为密切相关,根据RDDC数据库的统计(图4),RPE65基因目前已确认存在639个与LCA2相关的突变,其中127个被认定为致病性突变,这些突变已被广泛研究和报道。
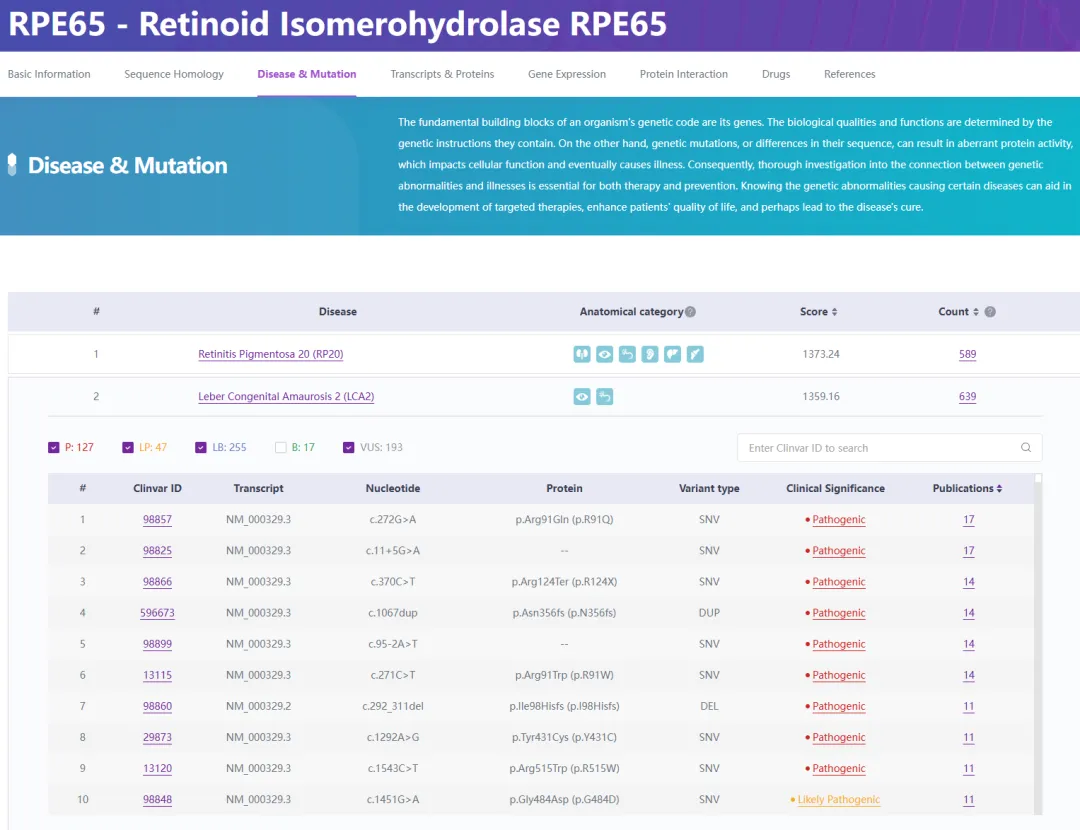
图4. RPE65基因突变与LCA2
来源:RDDC罕见病数据中心
了解更多疾病基因信息,欢迎登陆RDDC官网(https://rddc.tsinghua-gd.org),开启您的罕见病研究AI之旅。
疾病模型
动物模型的使用成为了不可或缺的工具。这些模型包括了我们熟知的斑马鱼、小鼠、大鼠、猫、犬以及鸟类等,它们各自独特的生物学特性为LCA的研究提供了丰富的实验平台。
斑马鱼作为一种在遗传学研究中广泛应用的模式生物,其眼睛结构与人类具有高度的相似性,使得斑马鱼成为研究视网膜发育和疾病机制的理想模型。特别是在模拟LCA相关的视网膜病变方面,斑马鱼模型能够精准地重现疾病过程,为研究疾病的发病机制提供了直观的证据。
犬类模型在LCA研究中同样占据了重要地位。相较于其他动物,犬类的寿命较长,这使得研究人员能够长期观察疾病的进展和演变,为评估治疗效果提供了更广阔的时间窗口。此外,犬类模型还允许研究人员在更接近自然环境下研究LCA,从而得到更为贴近实际的实验结果。
在LCA研究中,小鼠模型更是发挥着举足轻重的作用。由于小鼠在遗传和生理上与人类具有高度相似性,因此小鼠模型能够非常准确地模拟人类LCA患者的基因突变和病理特征。借助现代基因编辑技术,如CRISPR-Cas9,科学家们能够精确地在小鼠身上模拟人类LCA患者的基因突变,并观察这些突变如何影响视网膜的结构和功能。这些小鼠模型不仅展示了与人类LCA患者相似的病理特征,如光感受器细胞的逐渐退化、视觉功能的损失以及视网膜的病变,还为评估潜在的治疗方法提供了实验基础。为了更深入地理解LCA的发病机制和开发潜在的治疗方法,研究人员已经建立了多种小鼠模型来模拟这一疾病(图5)。
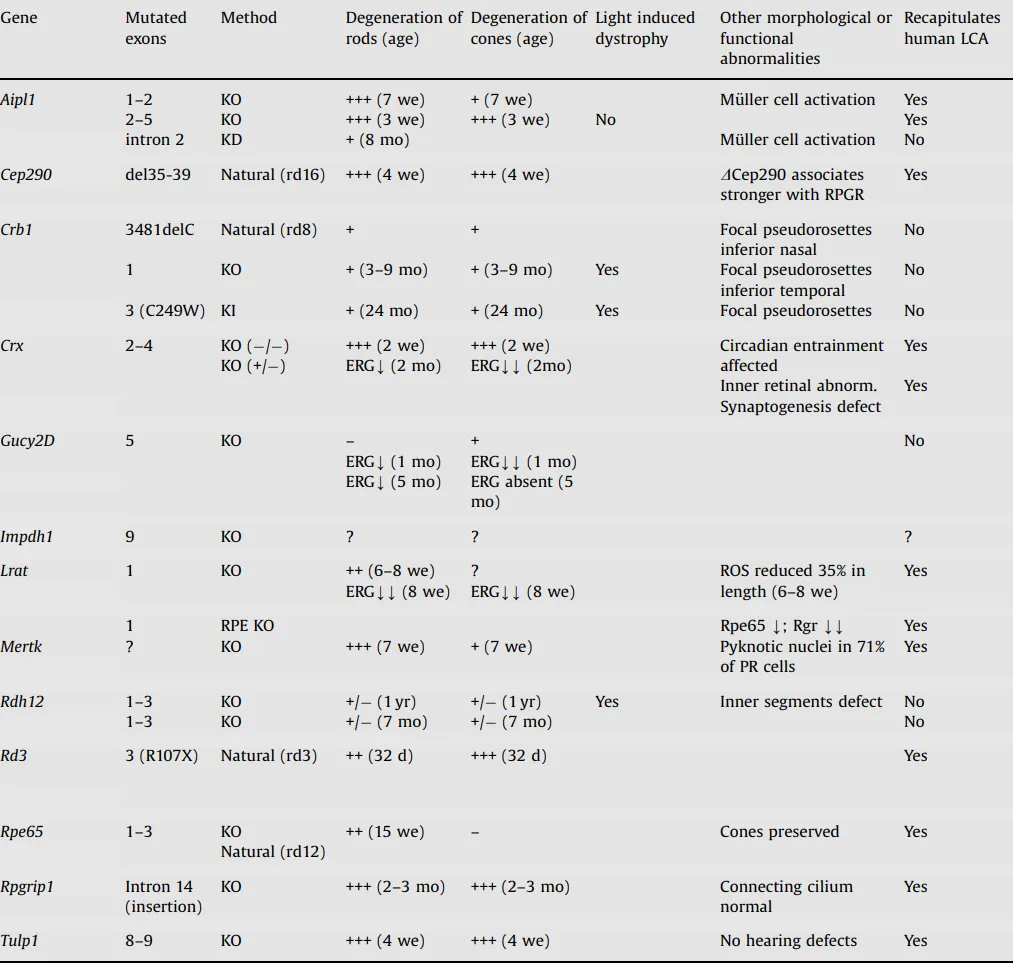
图5. 用于LCA的小鼠模型[13]
不同的小鼠模型各具特点:
Cep290敲除小鼠:这些小鼠缺乏CEP290基因,模拟了人类LCA患者中CEP290突变导致的视网膜病变。这些小鼠的视网膜发育不全,感光细胞逐渐退化,最终导致视力丧失;
Gucy2d敲除小鼠:这种小鼠模型模拟了GUCY2D基因突变导致的LCA。由于GUCY2D是光传导过程中的关键酶,这些小鼠的感光细胞在光刺激下无法产生正常的信号,导致视力受损;
RPE65敲除小鼠由于缺乏或异常的RPE65酶活性,无法有效将全反式视黄醛转化为11-顺式视黄醛,导致感光细胞的功能受损。这种功能损害进而引发视网膜病变,包括视力下降、夜盲症和视网膜色素变性等症状[13]。
通过这些小鼠模型,研究人员能够更直接地观察LCA的病理过程,并测试潜在的治疗方法。这些模型为LCA的研究和治疗提供了重要的工具。
治疗方法
基因增强疗法是LCA潜在治疗的主要来源。已经对常见涉及的基因进行了动物研究,包括GUCY2D、RPE65、AIPL1、RPGRIP1、LCA5、CEP290和RDH12,主要是通过使用腺相关病毒(adeno-associated virus, AAV)载体介导或慢病毒载体介导的基因增强疗法[6],载体的注射途径如图6所示,注射视网膜下或玻璃体内[14]。
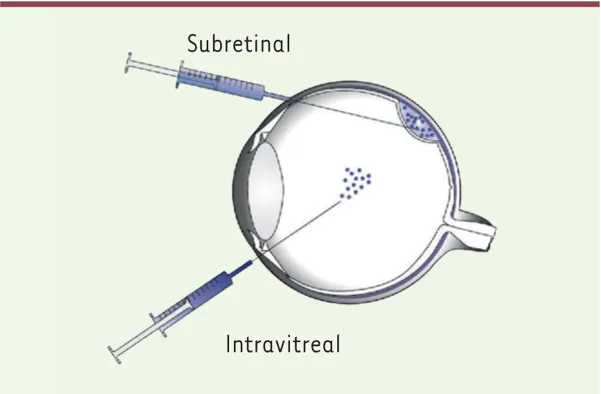
图6.基因治疗载体注射途径[14]
特别值得一提的是,由RPE65基因突变引起的LCA2具有其独特性。在此疾病中,视网膜神经感觉细胞在生命早期的保全尤为重要,因为基因治疗的核心思想是通过引入功能性野生型RPE65基因来替换受损基因,进而维持相对完整的视觉功能[15]。
根据智慧芽新药情报库中的LCA基因治疗药物统计(图7)可知:目前,针对RPE65突变的基因治疗已取得显著进展,其中Voreligene Neparvovec已进入上市阶段。此外,针对GUCY2D、RPE65和CEP290的临床试验也正如火如荼地进行中,这些试验旨在进一步验证基因增强疗法在治疗LCA中的有效性和安全性。值得注意的是,多个国家都积极参与到LCA基因治疗药物的研发中。在中国,辉大(上海)生物科技有限公司和启昇(上海)生物科技有限公司等公司也在该领域取得了重要进展。其中,辉大(上海)生物科技有限公司研发的药物已进入临床1/2期试验,而启昇(上海)生物科技有限公司的药物则已进入临床前阶段,这些进展为LCA患者带来了新的治疗选择和希望。
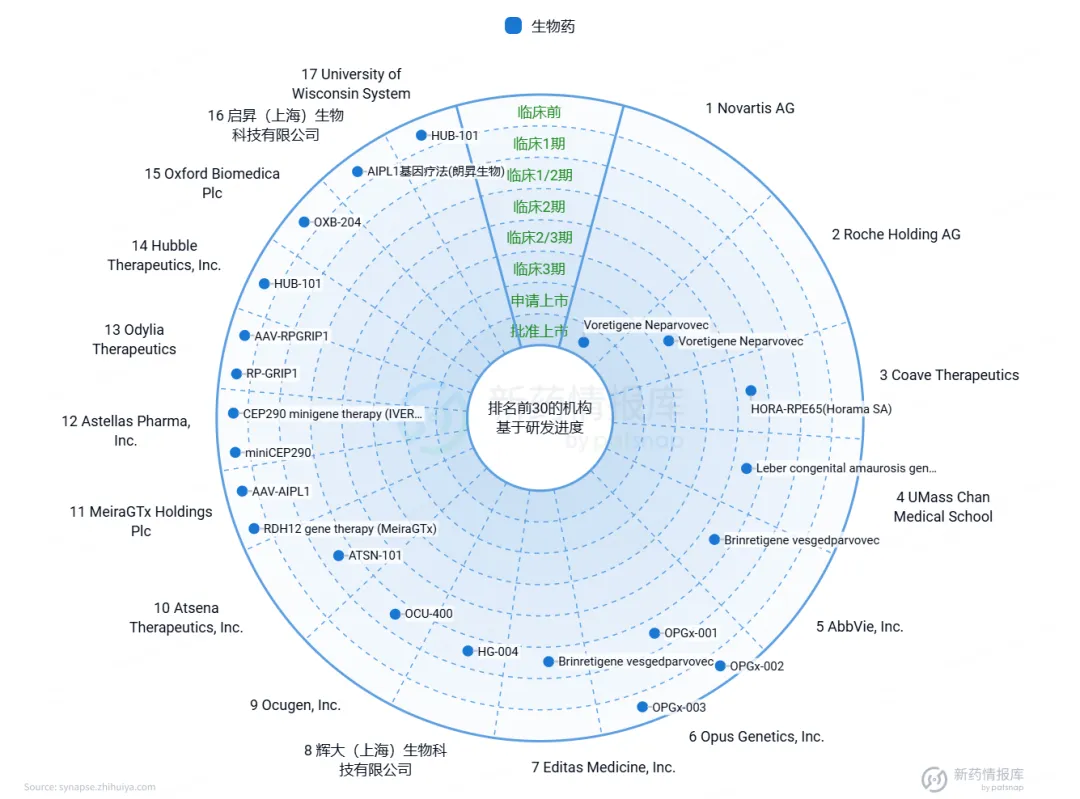
图7.LCA基因治疗药物研发现状
来源:智慧芽新药情报库
RDDC助力罕见病研究
罕见病数据中心(Rare Disease Data Center,简称“RDDC”),是由联盟内成员单位合作开发的关于罕见病研究的数据库。RDDC整合了全球开源的流行病学、药物研发、疾病相关基因图谱、基因突变位点、大小鼠实验动物模型等数据信息,再结合人工智能、生物信息等先进技术,部署了致病性预测工具(Pathogenicity Predictor)、RNA剪接预测模型(RNA Splicer)、通路分析(Pathway Analysis)等一系列AI、生信工具,赋能罕见病诊疗研究工作。
欢迎感兴趣的用户复制链接(https://rddc.tsinghua-gd.org)体验RDDC网站。

声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考资料:
[1]T.C. Chen, D.S. Huang, C.W. Lin, C.H. Yang, C.M. Yang, V.Y. Wang, J.W. Lin, A.C. Luo, F.R. Hu, P.L. Chen, Genetic characteristics and epidemiology of inherited retinal degeneration in Taiwan, NPJ Genom Med 6(1) (2021) 16.
[2]I. Perrault, J.M. Rozet, S. Gerber, I. Ghazi, C. Leowski, D. Ducroq, E. Souied, J.L. Dufier, A. Munnich, J. Kaplan, Leber congenital amaurosis, Mol Genet Metab 68(2) (1999) 200-8.
[3]R.K. Koenekoop, I. Lopez, A.I. den Hollander, R. Allikmets, F.P. Cremers, Genetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutions, Clin Exp Ophthalmol 35(5) (2007) 473-85.
[4]E.M. Stone, Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture, Am J Ophthalmol 144(6) (2007) 791-811.
[5]R.K. Koenekoop, An overview of Leber congenital amaurosis: a model to understand human retinal development, Surv Ophthalmol 49(4) (2004) 379-98.
[6]N. Kumaran, A.T. Moore, R.G. Weleber, M. Michaelides, Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions, Br J Ophthalmol 101(9) (2017) 1147-1154.
[7]K.L. Heher, E.I. Traboulsi, I.H. Maumenee, The natural history of Leber's congenital amaurosis. Age-related findings in 35 patients, Ophthalmology 99(2) (1992) 241-5.
[8]S.G. Foxman, J.R. Heckenlively, J.B. Bateman, J.D. Wirtschafter, Classification of congenital and early onset retinitis pigmentosa, Arch Ophthalmol 103(10) (1985) 1502-6.
[9]M. Alkharashi, A.B. Fulton, Available Evidence on Leber Congenital Amaurosis and Gene Therapy, Semin Ophthalmol 32(1) (2017) 14-21.
[10]S. Hanein, I. Perrault, S. Gerber, G. Tanguy, F. Barbet, D. Ducroq, P. Calvas, H. Dollfus, C. Hamel, T. Lopponen, F. Munier, L. Santos, S. Shalev, D. Zafeiriou, J.L. Dufier, A. Munnich, J.M. Rozet, J. Kaplan, Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis, Hum Mutat 23(4) (2004) 306-17.
[11]S.R. Dharmaraj, E.R. Silva, A.L. Pina, Y.Y. Li, J.M. Yang, C.R. Carter, M.K. Loyer, H.K. El-Hilali, E.K. Traboulsi, O.K. Sundin, D.K. Zhu, R.K. Koenekoop, I.H. Maumenee, Mutational analysis and clinical correlation in Leber congenital amaurosis, Ophthalmic Genet 21(3) (2000) 135-50.
[12]Y. Chen, Q. Zhang, T. Shen, X. Xiao, S. Li, L. Guan, J. Zhang, Z. Zhu, Y. Yin, P. Wang, X. Guo, J. Wang, Q. Zhang, Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis, Invest Ophthalmol Vis Sci 54(6) (2013) 4351-7.
[13]A.I. den Hollander, R. Roepman, R.K. Koenekoop, F.P. Cremers, Leber congenital amaurosis: genes, proteins and disease mechanisms, Prog Retin Eye Res 27(4) (2008) 391-419.
[14]J.B. Ducloyer, G. Le Meur, T. Cronin, O. Adjali, M. Weber, [Gene therapy for retinitis pigmentosa], Med Sci (Paris) 36(6-7) (2020) 607-615.
[15]S.G. Jacobson, T.S. Aleman, A.V. Cideciyan, A. Sumaroka, S.B. Schwartz, E.A. Windsor, E.I. Traboulsi, E. Heon, S.J. Pittler, A.H. Milam, A.M. Maguire, K. Palczewski, E.M. Stone, J. Bennett, Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success, Proc Natl Acad Sci U S A 102(17) (2005) 6177-82.