

国内首例!北京儿童医院报告一例 Curry⁃Jones 综合征
近日,首都医科大学附属北京儿童医院于《中华皮肤科杂志》报道一例 Curry⁃Jones 综合征,这也是国内首例确诊的 Curry⁃Jones 综合征。

据论文报告,2021 年 9 月 21 日,一名 3 岁 6 个月患儿因「多发带状白色斑疹」前往北京儿童医院皮肤科就诊,据家长描述,患儿出生后即发现身体躯干及肢体多部位皮肤出现条状、片状白色斑片,右手拇指较左手扁平,且右手活动度差。随年龄增长,皮疹亦随身体等比例增长,且右手拇指畸形和活动受限愈发明显。除此之外,患儿平时不爱与人交流,注意力也较差。患儿足月出生、发育正常,父母否认家族先天性疾病史,患儿母亲前 2 次怀孕均流产。
北京儿童医院皮肤科考虑不能排除以皮肤病变为重要表现的系统性先天性疾病,在首诊时即为患儿进行了全面体格检查,主要的发现有:
神经精神检查发现患儿精神状态正常,但交流时语言表达欠流畅,智力发育稍低于同龄儿童;
面部检查发现患儿存在特殊面容,包括眼距宽,睑裂窄,鼻梁扁平,反颌等特征,肢体检查发现右手拇指关节较左侧略膨大;
皮肤科检查发现患儿右胸部、右上肢屈侧、下腹正中线及右下肢可见多发片状、带状不规则的白色斑疹,大致沿 Blaschko 线呈条带状分布,局部略隆起;右手掌面、拇指桡侧可见条带状褐色斑。
根据上述临床表现,作者疑诊伊藤色素减少症(Hypomelanosis of Ito),故行进一步辅助检查。
患儿双手 X 光片示双手骨质未见明显异常,头颅 MRI 提示胼胝体发育不全。针对下肢白斑组织进行的皮肤活检发现:表皮基底层黑素细胞未见减少,真皮浅层基底样细胞呈团块样增生,部分与表皮相连,未见成熟毛囊结构,表现为基底样毛囊错构瘤。
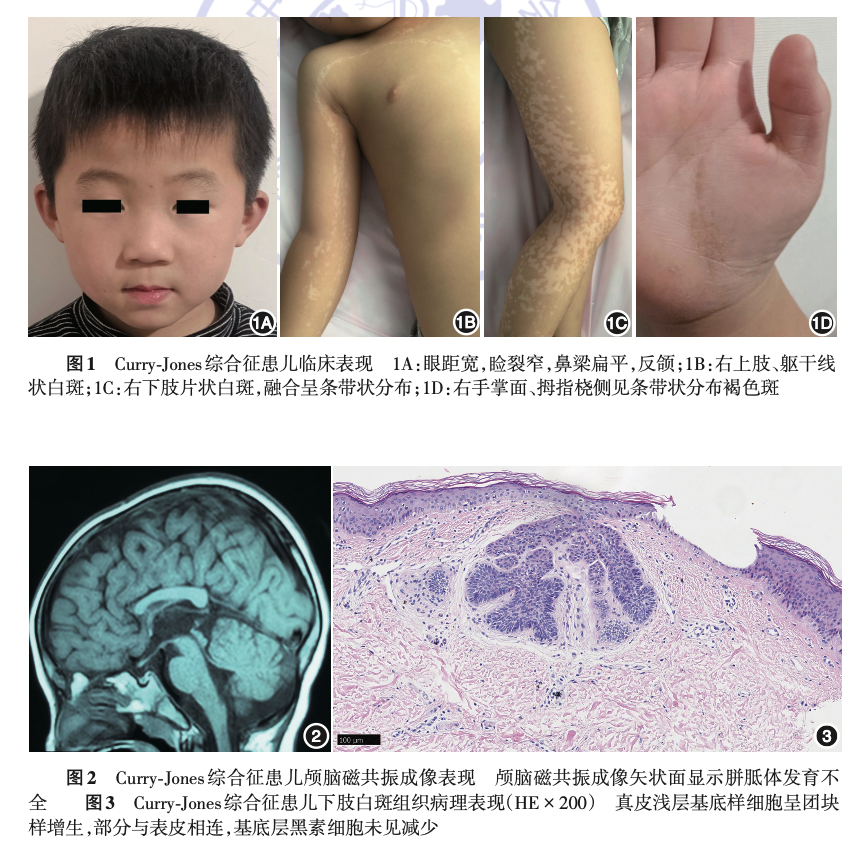
图2 来源:《Curry-Jones综合征1例国内首报》
因上述检查结果提示系统性先天性疾病的可能性进一步增加,在患儿家属知情同意下,取患儿皮损组织、静脉血和父母静脉血分别行基因测序。
经测序发现,患儿皮损组织 SMO 基因存在 c.1234C>T(p.L412F)突变,而患儿外周血和父母外周血 SMO 基因未见突变,结合临床发现,确诊其患有 Curry⁃Jones 综合征。
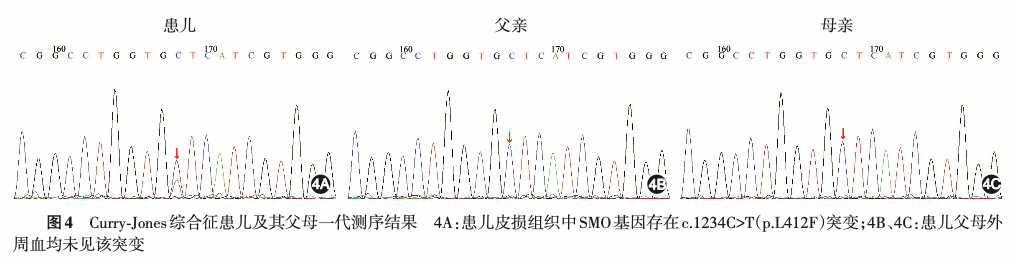
图3 来源:《Curry-Jones综合征1例国内首报》
Curry⁃Jones 综合征(Curry⁃Jones syndrome)是一种罕见的以带状皮肤色素异常、颅脑和肢端多种畸形及其他畸形或肿瘤等为特征的散发性先天性疾病,由重要的胚胎发育相关基因 SMO 的镶嵌突变引起。据不完全统计,此前全球范围有 13 例报道,且无中国本土病例。
Curry⁃Jones 综合征的主要表现为皮肤病变、发育畸形(可累及颅面部、神经系统、肢端骨骼系统和消化道系统等)和早发/多发肿瘤等,可通过典型的皮肤表现、特殊面容与皮损病理检查结果初步识别。
该病与伊藤色素减少症同为镶嵌突变相关疾病[3],且皮损、神经系统发育异常和骨骼畸形等临床表现较难区分,但后者的病变组织细胞常伴染色体畸形,且基因检测通常可显著区分两种疾病。鉴于镶嵌突变相关疾病的细胞遗传学特征,疑诊此类疾病时应取患者未受累部位(如外周血)和受累部位活检标本同时行检测,以免漏诊。
患儿目前定期随访。至末次随访时间 2022 年 11 月 9 日,患儿除皮损外无其他不适,发育尚可,右手拇指活动度仍欠佳,正行进一步右手康复训练中。
内容采集于丁香园
【声明】本文为转载文章,本平台仅作分享、传递信息,版权归原作者所有,如有侵权,请联系删除。