

文献分享 | 世界首次子宫内成功治疗遗传性罕见病:脊髓性肌肉萎缩症
一、研究概述
2025年2月19日,美国圣犹大儿童研究医院实验神经治疗中心主任Richard S. Finkel博士在The New England Journal of Medicine发表标题为“Risdiplam for Prenatal Therapy of Spinal Muscular Atrophy”的通信论文。该研究主要探讨了通过口服小分子药物Risdiplam对胎儿进行脊髓性肌肉萎缩症(SMA)的产前治疗。
研究中,一位因家族遗传背景被诊断为1型SMA的胎儿在孕32周5天至38周6天期间,通过母亲口服Risdiplam进行治疗,并在出生后持续用药至30个月。结果显示,治疗后胎儿未出现SMA的典型症状,SMN蛋白水平增加,神经丝水平降低,表明药物成功作用于靶点并影响了运动神经元的发育。
该研究由Finkel教授牵头,联合美国奥古斯塔妇产科合作伙伴(OBGYN Partners of Augusta)的JulieAnn Parker博士以及瑞士罗氏制药公司(F. Hoffmann–La Roche)的Heidemarie Kletzl博士共同完成。

Risdiplam(商品名:Evrysdi)是一种用于治疗脊髓性肌萎缩症(SMA)的小分子药物,由罗氏(Roche)旗下基因泰克(Genentech)公司与PTC Therapeutics联合开发。它通过调节SMN2基因的mRNA剪接,增加能够表达正常SMN蛋白的mRNA水平,从而提高SMN蛋白的生成,缓解SMA患者的症状。
药物特点- 作用机制:Risdiplam是一种SMN2前mRNA剪接修饰剂,能够增加SMN蛋白的产生,这种蛋白对维持健康的运动神经元和核心功能至关重要。- 给药方式:该药物可通过口服溶液或片剂形式给药,适合不同年龄段的患者。- 适应症:Risdiplam适用于2个月及以上儿童和成人SMA患者,涵盖1型、2型和3型SMA。
临床研究与应用- 临床试验:Risdiplam在多项全球多中心临床试验中表现出良好的疗效和安全性,包括FIREFISH(针对1型SMA婴儿)、SUNFISH(针对2型和3型SMA患者)和RAINBOWFISH(针对无症状SMA婴儿)等研究。- 产前治疗:2025年2月,《新英格兰医学杂志》报道了全球首例在胎儿期通过母亲口服Risdiplam治疗SMA的案例。该研究显示,产前治疗能够显著提高SMN蛋白水平,改善神经元功能。- 批准与应用:Risdiplam于2020年首次获得FDA批准,于2021年6月在中国获批上市。目前已在100多个国家获批,超过16,000名患者接受了治疗。
优势与意义- 非侵入性:作为唯一的非侵入性疾病缓解疗法,Risdiplam为SMA患者提供了便利。- 早期干预:产前治疗的成功案例为SMA的早期干预提供了新思路,可能改善患者的长期预后。
Risdiplam的开发和应用为SMA患者带来了新的希望,其在产前治疗中的成功应用更是开创了罕见病治疗的新纪元。
二、正文
Risdiplam是一种小分子药物,通过调节SMN2基因的剪接,增加脊髓性肌肉萎缩症(SMA)患者体内SMN(运动神经元存活)蛋白的水平,并改善疾病表现。一名胎儿因有一名已故同胞被遗传学确诊为1型SMA,而处于1型SMA的高风险中,通过羊水穿刺检测了SMA。检测结果显示,该胎儿SMN1基因无拷贝(确认SMA诊断),SMN2基因有两拷贝(预示为1型SMA)。既往数据表明,risdiplam相关物质能够通过胎盘,支持了产前治疗的可行性。本研究报告了对该胎儿使用risdiplam进行治疗的情况。
美国食品药品监督管理局和当地机构审查委员会批准了该单例患者的试验方案。在与独立倡导者的协商下,父母提供了书面知情同意。F. Hoffmann–La Roche公司提供了科学建议,就产前暴露的安全性提出了建议,并根据与赞助方(圣犹大儿童研究医院)的保密协议,免费提供risdiplam作为试验产品。从孕32周5天至38周6天分娩期间,母亲每天口服5 mg risdiplam。母亲每周接受产科健康和药物相关副作用的监测,胎儿通过超声检查接受生长、活动和解剖发育的监测。婴儿出生后第8天开始口服risdiplam,至今(2025年2月,婴儿30个月大)仍在持续每日给药。
在出生时,检测了母亲和婴儿的血液以及羊水中的risdiplam浓度、SMN和神经丝水平。母亲稳态时的risdiplam血浆谷浓度约为14 ng/ml。与母亲血浆浓度相比,分娩时羊水中的药物浓度为33%,脐带血中为69%。母亲和婴儿血样中SMN的水平,以及母亲和婴儿血浆样本中轻链和磷酸化重链形式神经丝的水平的连续测量结果总结于表1。
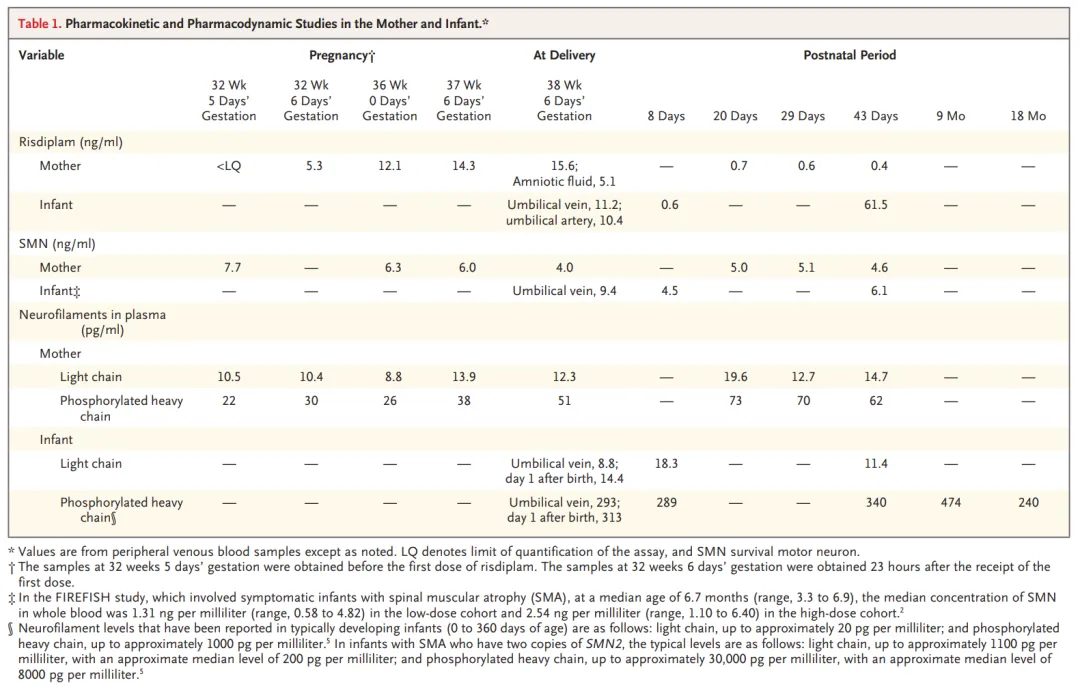
表1. 母婴药代动力学和药效学研究
婴儿出生时外观正常,但出生后被发现有因室间隔缺损引起的心脏杂音,该问题已自行解决。婴儿还存在轻度视力减退,伴有双眼视神经发育不良导致的短暂固定性眼震,以及与左侧中脑发育不良相关的轻度右侧偏瘫。到目前为止,婴儿还存在全面发育迟缓,但无倒退现象。微阵列和长读长基因组测序未发现其他遗传疾病,如与视隔发育不良和神经内分泌疾病相关的HESX1、OTX2和SOX2基因变异。
到目前为止,婴儿尚未出现SMA的特征性表现,如肌张力减退、肌无力、反射消失或肌束颤动。每6个月进行一次的运动功能、肌肉超声和电生理学研究显示,婴儿的周围神经和肌肉发育正常(详见NEJM.org上本信函全文的补充附录)。
本研究通过母亲口服给药的方式将risdiplam给予SMA胎儿,并持续给予儿童至30个月大,治疗与SMA症状的缺失相关。SMN水平的增加和神经丝水平的降低支持了该药物对靶点的参与以及对运动神经元发育的影响。研究人员认为,先天性异常可能发生在胎儿接触risdiplam之前的早期发育阶段,尚未找到原因;这些变化在产前和产后接受risdiplam治疗的动物中未被观察到。这一单一病例的结果不能被广泛推广,但可能支持对产前诊断为SMA的胎儿进行risdiplam治疗的考虑。
来源:BioMedDaily
【声明】本文为转载文章,本平台仅作分享、传递信息,版权归原作者所有,如有侵权,请联系删除。