

Hereditary Angioedema (HAE) [1] - A Rare and Dangerous Disease
Disease Overview
Hereditary Angioedema (HAE) is a rare disease that many doctors may never encounter in their lifetime [1]. First described by Dr. Osler in 1888, HAE is a genetic disorder characterized by recurrent, self-limiting tissue swelling, particularly when subjected to minor trauma or increased mental stress. Triggers causing the swelling are often unknown to most patients. HAE is an autosomal dominant inherited disease.
Though rare in clinical practice, HAE can easily be misdiagnosed [2]. In May 2018, the National Health Commission and other departments included HAE in the "First Batch of Rare Diseases Catalog" [3].

Figure 1. Sourced from West China Hospital of Sichuan University
Epidemiology
Currently, the global prevalence of HAE is between one in 50,000 to one in 100,000. Based on this prevalence, China has an estimated 30,000 HAE patients [4], but only about 500 diagnosed cases, giving a diagnosis rate of less than 2%. There are fewer than 100 doctors in the country who are aware of and capable of diagnosing HAE, mainly concentrated in top-tier hospitals in major cities such as Beijing, Shanghai, and Guangzhou.
As an autosomal dominant hereditary disease, if one parent has HAE, their children have a 50% chance of inheriting the disease. Research shows that 75% of HAE cases are familial, while 25% are caused by gene mutations [5].
Pathogenesis
Hereditary angioedema (HAE) primarily presents as recurrent swelling of soft tissues such as the skin and/or mucous membranes and is based on molecular genetics of C1 esterase inhibitor (C1-INH) or FⅫ gene mutations [2]. The understanding of HAE's pathogenesis in the medical community has deepened with the continuous advancement in the field of molecular genetics, especially in research on C1-INH.
HAE can be mainly categorized into the following types:
1. Type I (Classical): HAE with decreased C1-INH levels and function due to mutations in the SERPING1 gene located on chromosome 11q11-13.1. The Human Gene Mutation Database has reported over 450 different mutations in the C1-INH gene region as of mid-2018.
2. Type II (Variants): HAE with normal or elevated C1-INH levels but functionally impaired protein due to mutations in the SERPING1 gene.
3. Type III: HAE with normal levels of C1-INH, caused by mutations in the angiopoietin-1 gene (ANGPT1) or other genes, resulting in increased activation of factor XII and the contact system.
4. HAE with unknown pathogenic gene.
C1-inhibitor (C1-INH) is a serine protease inhibitor present in human plasma and is a critical regulatory factor in the complement, fibrinolytic, coagulation, and kinin-forming systems. It is primarily synthesized in the liver. In 2002, Han et al. first demonstrated through animal experiments that bradykinin (BK) is the predominant mediator in the pathophysiology of HAE. The deficiency of the regulatory protein C1-INH in the complement system impairs control over the contact activation system. Upon activation of the contact system, clotting Factor XII and plasma kallikrein (PKK) mutually cleave each other, producing active proteases FXIIa and PKK. PKK then cleaves high-molecular-weight kininogens to release bradykinin. Bradykinin binds to B2 receptors (BKB2R), triggering the release of nitric oxide (NO), prostacyclin, and endothelial-derived hyperpolarizing factors. These events result in vasodilation, increased vascular permeability, and smooth muscle contraction, ultimately leading to fluid exudation and vascular edema [6].
Clinical Manifestations
Hereditary angioedema is characterized by acute, recurrent swelling of subcutaneous and/or submucosal tissues, with non-pitting, self-limited, and localized characteristics. The symptoms and signs differ greatly between patients and even between each attack in the same patient, including variations in the age of onset, frequency, location, and severity. These differences may be related to environmental factors and gene mutation types, but research is currently lacking in this area [7].
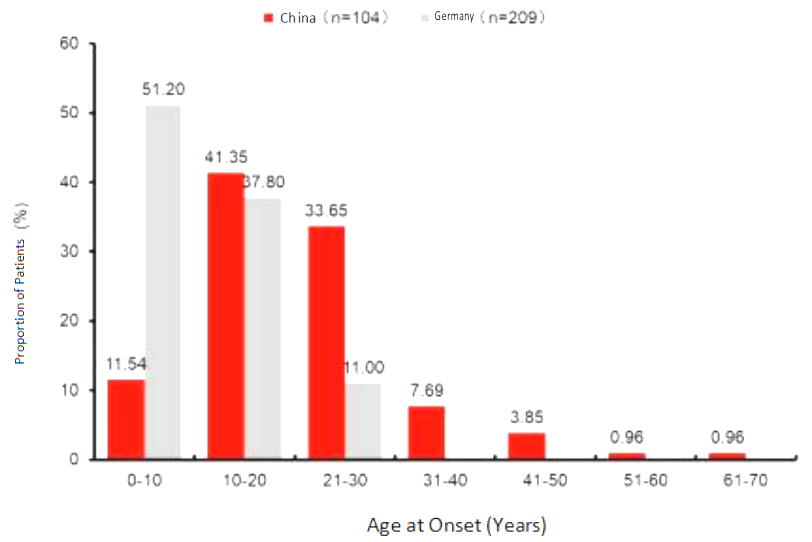
Figure 2. Age of onset for HAE patients.
The average age of onset for HAE patients in China is 21.25 years old (range: 2-63 years old), and most patients (75%) experience their first attack between the ages of 10 to 30 years old. Only 11.54% of patients experience the disease's onset before the age of 10. This contrasts with Germany, where about half of the patients (51.2%) experience the disease's onset before the age of 10. [8] The fact that most HAE patients experience their first attack between the ages of 10 to 30 years old, rather than at birth or during infancy, can lead to misconceptions that it is not a hereditary disease if symptoms appear later in life, thereby complicating the diagnosis process. The age range of 20-30 is a critical period for education and career development, and frequent swelling can cause significant disruptions to daily life and academic and career pursuits.
HAE symptoms can vary from person to person, with some adolescents potentially experiencing an increased frequency of attacks during puberty. Some people may feel a prodrome, such as a tingling sensation in the affected area, before swelling occurs. Swelling, involved in an attack, will typically intensify over several hours and then gradually recede over the next 48-72 hours if left untreated. An attack may also start in one area and then spread to another before resolving. Untreated throat swelling, the most dangerous symptom of HAE, can progress rapidly, resulting in difficulty breathing or suffocation. It carries a mortality rate of 11% to 40%, making it a leading cause of death among patients with this disease.
References:
[1] Osler W. Hereditary angio-neurotic oedema[J]. Am J Mcd Sci, 1970, 95:362-367.
[2] Li CX. Report on six cases of hereditary angioedema in one family[J]. Clinical Misdiagnosis & Mistreatment, 2017, 30(03):23-25. (In Chinese)
[3] National Health Commission, Ministry of Science and Technology, Ministry of Industry and Information Technology, National Medical Products Administration, and National Administration of Traditional Chinese Medicine. First Batch of Rare Diseases Catalog. National Health and Medical Development (2018) No.10. (In Chinese)
[4] Li S. New hope for patients with hereditary angioedema [N]. Health Times, 2023-02-28(015). (In Chinese)
[5] National Health Commission, Office of the Expert Committee on Diagnosis, Treatment, and Assurance of Rare Diseases (Peking Union Medical College Hospital). Guidelines for Diagnosis and Treatment of Rare Diseases (2019 Version). 2019;38–189. (In Chinese)
[6] Guan ZW, Li QF. Progress in prevention and treatment of hereditary angioedema[J]. Journal of Chinese and Western Dermatology, 2021,20(04):424-426.
[7] Cao Y, Liu S, Zhi YX. Research progress on pathogenesis of hereditary angioedema[J]. Journal of Chinese Academy of Medical Sciences, 2020, 42(05):686-690. (In Chinese)
[8] Xu YY, Zou XY, et al. Hereditary angioedema in Chinese patients: a need for awareness. Eur J Dermatol. 2013 Jul-Aug;23(4):500-4.