

Spinocerebellar Ataxia (SCA) [1] - The shaky 'Penguin Disease' caused by genetic disorder
Spinocerebellar Ataxia (SCA) [1] - The shaky 'Penguin Disease' caused by genetic disorder
Disease overview
A special couple appeared on an episode of the TV show "The Reader". After more than a year of efforts, a stubborn boy and a girl who couldn’t walk had used a tricycle and a wheelchair to create a touching track of love track across China. For ordinary people, leaving one's hometown requires courage, but for Lai, a woman with familial Spinocerebellar Ataxia (SCA), simply standing was a luxury. Ms. Lai wouldn’t let that stop her, "The world is so big, I want to go and see it," she said.
Figure 1. Source: Reproductive Medicine Center, First Affiliated Hospital of Soochow University.
Spinocerebellar ataxia (SCA), commonly known as Penguin Disease, is a group of autosomal dominant inherited diseases characterized by progressive cerebellar ataxia. The most common are the polyglutamine (polyQ) SCAs, which are caused by abnormal expansion of cytosine-adenine-guanine (CAG) repeats in the gene coding region. Pathologically, SCA mainly affects the cerebellum, leading to Purkinje fiber loss, and may also affect the spinal cord, brainstem, and peripheral nerves. The worldwide incidence rate ranges from 1/100,000 to 5/100,000, with regional variations. [1]
Figure 2. Source: Dizziness Treatment Center, Luohe Central Hospital
Pathogenesis
The pathogenesis of SCA is currently not fully understood. There are several possible mechanisms, which may be interrelated and overlapping, including:
1. Protein toxicity: Protein toxicity plays a central role in the pathogenesis of polyglutamine SCA. Mutant proteins are prone to conformational changes and aggregation, which can recruit normal proteins or molecular chaperones to form aggregates inside the cells, more likely to formpreferably in the nucleus. This disrupts protein homeostasis in the cells and leads to various downstream signaling pathway changes through multiple mechanisms. Some traditional mutations in non-polyglutamine expansion SCAs (such as SCA14, SCA35) can also cause misfolding of proteins and abnormal aggregation.
2. RNA toxicity: For non-coding region repeat expansion type SCAs, transcripts containing repeat sequences can form nuclear RNA clusters, which sequester RNA-binding proteins and interfere with splicing or other RNA-dependent processes, disrupting protein homeostasis and leading to cell toxicity.
3. Ion channel dysfunction: Pathogenic mutations in genes that encode ion channels or signaling pathway components that regulate channel activity can cause abnormal cerebellar neurophysiological signals or drive neuronal dysfunction.
4. Impaired bioenergetics: Mutant proteins can directly affect the structure and function of mitochondria (SCA28), or indirectly affect mitochondrial function through cellular pathways, such as transcriptional dysregulation mediated by polyglutamine SCA disease proteins, and autophagy/lysosomal pathways, thereby affecting the production of bioenergy.
5. Loss of nuclear integrity: Loss of nuclear integrity refers to the breakdown of both the structure and function of the nucleus, including gene expression dysregulation, DNA repair damage, and disruption of nuclear transport, among others.
6. Others: In recent years, a non-ATG-mediated RNA translation has been found in repeat expansion diseases, which forms easily aggregating polypeptides, and this translation exists in some SCAs. However, the role of RNA translation in SCA is still unclear.
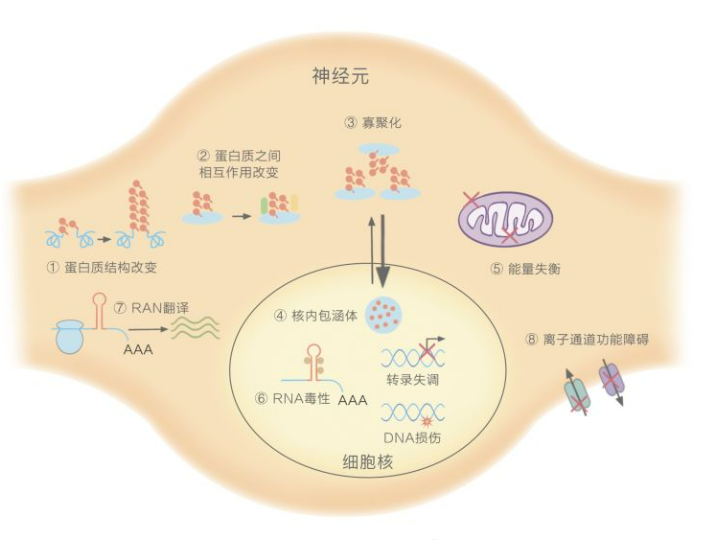
Figure 3. Pathological mechanisms underlying SCA [3]
Disease Cclassification
SCA is a genetically heterogeneous disease, with clinical manifestations that are extremely similar yet each has its own characteristics. Currently, the classification of SCA is based on genetic loci, and each subtype is named SCAn (with n progressing according to the time of discovery of the causative gene or locus), and more than 40 subtypes have been identified. The pathogenesis of SCA is not fully understood. Currently, there are multiple pathological mechanisms that may lead to neuronal degeneration in SCA.
1. Abnormal repeat numbers of polyglutamine chains: SCA1, 2, 3, 6, 7, 17, DRPLA. The expansion of repeat numbers can: 1) Change the protein's original structure and function; 2) Affect the interaction with other proteins; 3) Promote oligomerization, leading to protein toxicity; 4) Form inclusion bodies in the nucleus, causing transcriptional dysregulation, DNA damage, and disrupting nuclear integrity; 5) Cause energy imbalance in the cytoplasm, ultimately damaging or killing the entire neuron cell.
2. Abnormal repeat numbers inof non-coding sequences: SCA8, 12, 10, 31, 36, 37. The expansion of repeat numbers can: 6) Sequester critical RNA-binding proteins inside the nucleus, disrupting normal splicing and causing RNA toxicity; 7) Trigger repeat sequence-associated non-ATG-mediated translation in the cytoplasm, generating easily aggregating peptides and producing protein toxicity.
3. Ion channel gene variations: SCA6, 13, 19/22, 15/16, 29, 41, 42, 44. These mutations can: 8) Cause channel dysfunction, leading to cerebellar circuit disorders and resulting in ataxia. [4]
Polyglutamine SCA is the most common, and among the Han population in China, the frequency of different subtypes of SCA from high to low is SCA3, SCA2, SCA1, SCA6, SCA7, SCA12 [5].[5] The mutated genes and loci of SCA subtypes are present in different chromosomal intervals. Based on the gene loci and clinical phenotype characteristics, SCA can be classified as SCA1-SCA45, as detailed in Table 1.
Table 1. Names, causative genes, loci, and clinical characteristics of various SCA subtypes [5].[5]

The incidence and prevalence of SCA subtypes vary between different regions and races, and even among individuals of the same race worldwide. Currently, it is believed that this may be related to individual differences between different regions and races, or to the "founder effect" within populations.
Clinical manifestations
The clinical manifestation of SCA is a progressive ataxia mainly originating from cerebellar lesions but can also result from sensory and vestibular disturbances. Patients initially present with gait instability, and as the disease progresses, they may have difficulty writing (often manifested as "macrographia") and fine motor impairments. Almost all SCA patients experience speech disorders and swallowing problems. Eye symptoms can include nystagmus (vertical, horizontal, or mixed), slow saccadic eye movements, and diplopia, among others. In addition, SCA also has complex and diverse non-ataxia symptoms, including motor symptoms (spasms and atrophy), movement disorders (Parkinson's disease, dystonia, and chorea), eye movement disorders related to brainstem dysfunction (slow saccadic eye movement or gaze paralysis), sensory symptoms, epilepsy, myoclonus, cognitive and intellectual impairments, urination symptoms, and sleep disorders (including restless leg syndrome, rapid eye movement sleep behavior disorder, excessive daytime sleepiness, insomnia, and sleep apnea).
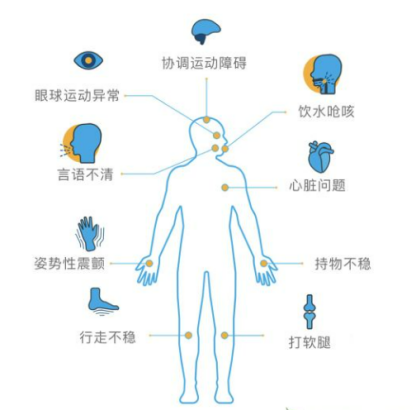
Figure 4. Clinical manifestations of SCA [6]
Most SCA patients have an onset age between 30 and 40 years old. Some non-polyglutamine expansion SCAs (such as SCA5, SCA21, etc.) or SCAs with a very large number of CAG repeats can onset in childhood. SCA caused by dynamic mutations has a negative correlation between the onset age and the length of repeats. Due to the instability of repeat expansions in somatic cells and germ cells, the successive generations of a family may experience a phenomenon called genetic anticipation, whereby the age of onset of the disease gradually advances, and the clinical phenotype worsens. The progression of SCA is generally evaluated through ataxia assessment and rating scales (such as SARA). SARA is a semi-quantitative assessment based on the level of ataxia impairment and has been extensively used worldwide for observational and interventional studies after rigorous verification. Cohort studies on SCA progression and prognosis have found that SCA1 progresses the fastest, SCA2 and SCA3/MJD progress are intermediate, and the progression of SCA6 is the slowest among the most common polyglutamine SCAs [2].
This article is a collection and summary of information. If there are any errors or omissions, please feel free to provide criticism and corrections!
Reference sources and image sources:
[1] Sun D, Hu XY. Progress in treatment research on polyglutamine-induced spinocerebellar ataxia. J Gen Pract Clin Educ. 2021;19(4):351-354. doi: 10.13558/j.cnki.issn1672-3686.2021.004.020.
[2] Wu FR, Zhong M. Clinical presentation, pathogenesis, diagnosis, and treatment of spinocerebellar ataxia. Shandong Medicine. 2021;61(23):112-115.
[3]“HEREDITARY ATAXIAS: DOMINANT.” NEUROMUSCULAR DISEASE CENTER, Washington University, http://neuromuscular.wustl.edu/ataxia/domatax.html.
[4]https://zhuanlan.zhihu.com/p/345416569
[5] Li SL, Zhai XJ, Zhang L, Yan ZY, Xie LX, Han YL, Lü JJ, Chen WW, Guo XQ, Dong H. Pathogenesis and clinical phenotype of hereditary spinocerebellar ataxia. Journal of Henan University (Medical Science). 2018;37(4):295-300. doi: 10.15991/j.cnki.41-1361/r.2018.04.018.
[6]“What Is Ataxia?” National Ataxia Foundation, http://ataxia.org/what-is-ataxia.