

遗传性脊髓小脑共济失调 (SCA)[1]——摇摇晃晃的“企鹅病”
疾病概述
《朗读者》节目有一期迎来了一对特殊的嘉宾。一个倔强的男孩,一个走不动路的姑娘,历时一年多的时间,用一辆三轮车和一架轮椅在中国大地上勾勒出了一幅感人的爱心足迹。“世界那么大,我想去看看”,对于普通人来说,走出去需要的是勇气,但是对于身患家族遗传性小脑共济失调症的赖女士来说,简单的站立对她来说都是一种奢望。
图片来源于苏州大学附属第一医院生殖医学中心
遗传性脊髓小脑共济失调 (spino cerebellar ataxia,SCA) 俗称企鹅病,是一组常染色体显性遗传疾病,临床特征是进行性共济失调,其中最常见的是多聚谷氨酰 (polyglutamine,polyQ) SCAs,它们是由于基因编码区的胞嘧啶-腺嘌呤-鸟嘌呤 (cytosine-adenineguanine,CAG) 异常重复扩增导致的。SCA病理上主要累及小脑,常伴有浦肯野纤维丢失,也可累及脊髓、脑干及周围神经,全球发病率为1/100000-5/100000,不同地区具有差异[1]。
图片来源于漯河市中心医院眩晕诊疗中心
发病机制
SCA的发病机制目前尚不明确,主要存在以下几种可能的机制,相互之间可能有相互关联、重叠:
1.蛋白毒性:蛋白毒性在多聚谷氨酰胺SCA的发病机制中具有中心作用,突变型蛋白容易构象异常并聚集,可招募正常的蛋白或分子伴侣在细胞内共同形成聚集体(更倾向于在核内形成),通过多种机制使细胞内的蛋白平衡破坏而产生不同的下游信号通路改变。另一些非重复扩展型SCA (如SCA14、SCA35) 中的传统突变也可导致蛋白的错误折叠而异常聚集。
2.RNA毒性:对于非翻译区重复扩张型SCA,含有重复序列的转录本形成核内RNA团簇,这些团簇隔离RNA结合蛋白,干扰剪接或其他依赖RNA的过程,甚至影响细胞内蛋白平衡,导致细胞毒性。
3.离子通道功能障碍:编码离子通道或调节通道活性信号通路组件的基因致病性突变,会导致小脑神经电生理信号异常或驱动神经元功能障碍。
4.生物能量受损:突变型蛋白可直接影响线粒体的结构和功能 (SCA28),也可通过如多聚谷氨酰胺SCA疾病蛋白介导的转录失调、自噬/溶酶体通路等细胞途径间接影响线粒体功能,从而影响生物能量产生。
5.核完整性丧失:核完整性丧失是指细胞核结构和功能均受到破坏,其中包括基因表达失调、DNA修复受损、核浆运输被破坏等。
6.其他:近年来,在重复扩张疾病中发现一种非ATG介导的RNA翻译,形成易于聚集的多肽,而这种翻译存在于一些SCA中,但RNA翻译在SCA中存在何种作用尚不明确[2]。
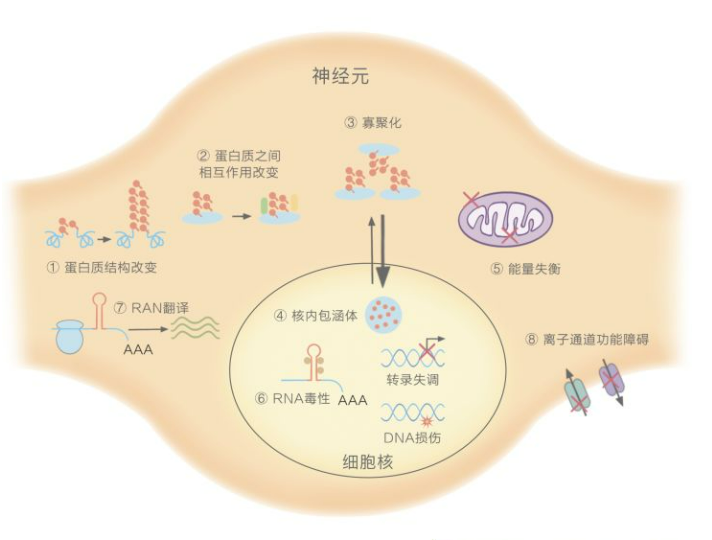
SCA的病理生理学机制[3]
疾病分类
SCA是遗传异质性疾病,各亚型临床表现极为相似又各有特征,目前倾向于根据遗传位点对SCA进行分类,每种亚型都被命名为SCAn (n依照致病基因或位点发现的时间顺序递进),目前已发现超过40种亚型。SCA的发病机制尚未完全阐明,目前, SCA存在多种可能导致神经元退化的病理生理学机制:
1.多聚谷氨酰胺链的重复数量异常:SCA1,2,3,6,7,17,DRPLA。①改变蛋白质原本的结构与功能;②影响与其他蛋白质的相互作用;③倾向于寡聚化,产生蛋白毒性;④在细胞核内,形成包涵体,引起基因转录失调、DNA损伤,破坏核完整性;⑤在细胞质中,引起能量失衡,最终造成整个神经元细胞的损伤甚至死亡。
2.非编码区的重复序列数量异常:SCA8,12,10,31,36,37。⑥在细胞核内,与关键的RNA结合蛋白螯合,干扰正常剪接,产生RNA毒性;⑦在细胞质中,引发重复序列相关的非ATG介导的翻译,生成易于聚集的多肽,产生蛋白毒性。
3.离子通道基因变异:SCA6,13,19/22,15/16,29,41,42,44。⑧导致通道功能障碍,引起小脑回路紊乱,造成共济失调[4]。
多聚谷酰胺SCA最为常见,在我国汉族人群中SCA各亚型出现的频率从高到低依次为SCA3、SCA2、SCA1、SCA6、SCA7、SCA12[5]。SCA各亚型突变基因及其位点在不同的染色体区间上,根据基因位点及临床表型特点可将SCA分为SCA1-SCA45,详见表1。
表1 SCA各表型名称、致病基因、位点及临床特点[5]

在世界范围内不同区域和不同种族间,甚至在同一种族内,SCA的发病频率和各个表型的发病率各有差异。目前认为可能和不同区域、不同种族的个体差异或“奠基人效应 (founder- effect)”相关。
临床表现
SCA的临床表现是进行性共济失调,主要源于小脑病变,但也可由感觉性和前庭性引起。患者首先表现为步态不稳,随着病情进展,出现书写困难 (常表现为“书写过大症”) 及精细运动障碍,几乎所有的SCA患者会出现言语障碍及吞咽问题。眼部症状可表现为眼震 (垂直性、水平性或混合性)、眼球扫视运动和追踪运动缓慢、复视等。此外,SCA还存在复杂多样的非共济失调症状,包括运动症状 (痉挛和肌萎缩)、运动障碍 (帕金森病、肌张力障碍和舞蹈症)、与脑干功能障碍有关的眼球运动异常 (快速眼球运动减慢或凝视麻痹)、感觉症状、癫痫、肌阵挛、认知和智力障碍、泌尿症状及睡眠障碍 (包括不宁腿综合征、快速眼动睡眠行为障碍、日间过度睡眠、失眠和睡眠呼吸暂停)。
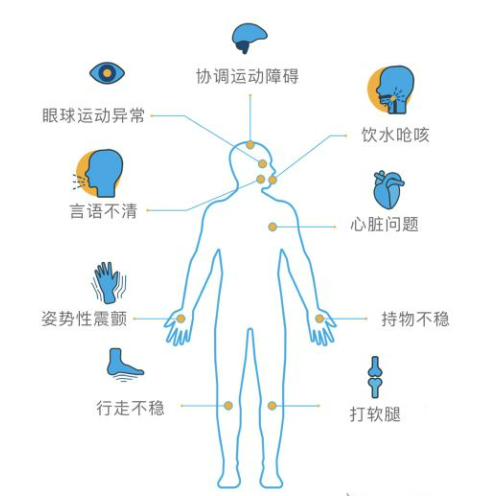
SCA临床表现[6]
大多数的SCA患者的发病年龄一般在第30-40岁之间。一些非重复扩展型SCA (如SCA5、SCA21等) 或CAG重复次数非常多的SCA可在儿童期发病。由动态突变引起的SCA,发病年龄与重复长度呈负相关关系。由于重复扩张在体细胞和生殖细胞中的不稳定性,家系中的连续世代会经历遗传早现现象,即后代发病年龄逐代提前、临床表型逐渐加重。SCA疾病进展一般通过共济失调评估和评级量表 (SARA) 来评测,SARA是一种基于对共济失调损伤水平的半定量评估的临床量表,经过了严格的验证,目前在世界范围内被广泛用于观察性和干预性研究。关于SCA进展及预后的队列研究发现,在最常见的多谷氨酰胺SCA中,SCA1进展速度最快,SCA2、SCA3/MJD进展速度中等,SCA6进展速度最慢[2]。
声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考文献及图片来源:
[1]孙迪,胡兴越.多聚谷氨酰胺脊髓小脑性共济失调的治疗研究进展[J].全科医学临床与教育,2021,19(04):351-354.DOI:10.13558/j.cnki.issn1672-3686.2021.004.020.
[2]吴方瑞,钟敏.脊髓小脑性共济失调的临床表现、发病机制及诊疗研究进展[J].山东医药,2021,61(23):112-115.
[3]“HEREDITARY ATAXIAS: DOMINANT.” NEUROMUSCULAR DISEASE CENTER, Washington University, http://neuromuscular.wustl.edu/ataxia/domatax.html.
[4]https://zhuanlan.zhihu.com/p/345416569
[5]李松林,翟小菊,张璐,闫中义,谢龙祥,韩亚莉,吕佳佳,陈文武,郭向前,董寰.遗传性脊髓小脑共济失调的发病机制及临床表型[J].河南大学学报(医学版),2018,37(04):295-300.DOI:10.15991/j.cnki.41-1361/r.2018.04.018.
[6]“What Is Ataxia?” National Ataxia Foundation, http://ataxia.org/what-is-ataxia.