

遗传性痉挛性截瘫(HSP)——“猞猁的耳朵”
疾病概述
有这样一种疾病患者脑MRI可见“猞猁耳”征和胼胝体变薄,这个疾病就是在遗传性痉挛性截瘫(HSP)的高度特导性影像学中发现的,胼胝体辐射线额部(forcens minor)(膝纤维)的T2(组织横向弛豫快慢)和T1(组织纵向弛豫快慢)弛豫时间延长,可能导致该区域的T1低信号和T2/FLAIR(磁共振成像液体衰减反转序列)高信号,由此可以观察到貌似猞猁耳朵顶部的簇手[1]。
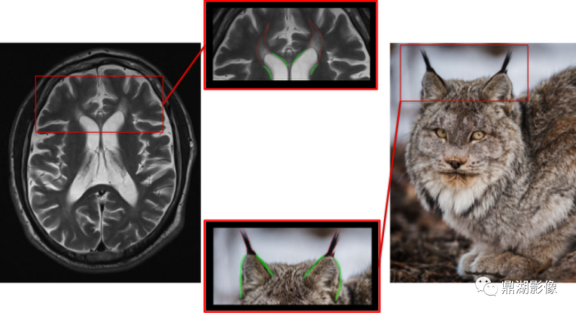
图1 遗传性痉挛性截瘫患者MRI影像 图源来自于movementdisorders
遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP),又称Strümpell-Lorrain病,是一组具有显著临床和遗传学异质性的神经退行性疾病。HSP主要的临床特征表现为双下肢缓慢进行性无力和痉挛性截瘫,发病率约为1.8/100000[2-3]。
图2 遗传性痉挛性截瘫患者 图源来自于腾讯医典
发病机制
HSP主要病理改变为皮质脊髓束及后柱最长的下行性运动纤维远端轴索的变性,伴或不伴脱髓鞘改变,主要由囊泡转运、轴索运输、细胞器形成、线粒体功能异常、脂质代谢障碍、髓鞘异常等分子机制所致,以胸髓为重,其次为脊髓小脑束、薄束,脊髓前角、基底核、小脑、脑干、视神经也可受累。尽管HSP具有广泛的遗传和表型异质性,但在HSP发病中已经明确了几个普遍认可的主要原因:
① 膜转运功能或内质网形态异常;
② 轴浆运输异常;
③ 髓鞘形成异常;
④ 线粒体蛋白质异常;
⑤ 脂质代谢紊乱。
微管动力学、轴突转运和线粒体功能的改变被认为是导致HSP远端神经退行性变的机制。虽然HSP被认为是一种上运动神经元病变,但有研究表明,HSP病人运动和感觉神经束在中枢和周围神经系统中的病变更为广泛。电生理异常的分布模式与不同的HSP基因型相关,可以反映不同的潜在病理机制[4]。
去年,中国科学院脑科学与智能技术卓越创新中心(神经科学研究所)、神经科学国家重点实验室研究员刘静宇研究组通过对遗传病家系的连锁分析和Sanger测序,并结合细胞系模型分析,发现遗传性痉挛性截瘫(HSP)致病基因SPAST编码的截短蛋白质spastin可以通过异构体特异性方式干扰微管的动态平衡,进而导致HSP的发生。此研究进一步提出spastin的截短突变体可能通过长期的细胞积累方式影响皮质脊髓束的功能,为探究遗传性痉挛性截瘫4型(SPG4)的致病机制和开展治疗提供了新的方向[5]。
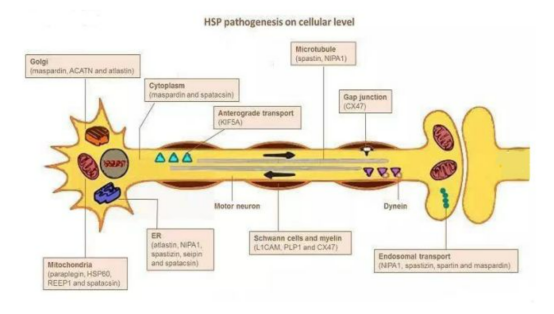
图3 遗传性痉挛性截瘫细胞水平的发病机制[6]
疾病分类
HSP主要根据临床表现、遗传方式及突变基因进行分类。根据临床表现,HSP分为单纯型和复杂型:
单纯型HSP特征为表现为逐渐进展的双下肢痉挛、步态不稳、腱反射亢进,可合并膀胱括约肌功能障碍、踝关节振动觉减退。
复杂型HSP除了单纯型HSP表现出的特征外,还可伴有智力障碍、锥体外系症状、共济失调、癫痫、白内障、视神经萎缩、视网膜变性、鱼鳞病及周围神经病等。
目前已证实有近60种不同的基因位点与HSP有关,其中:
19种为常染色体显性遗传的痉挛性截瘫(AD-HSP),40%~45%的患者携带SPAST基因突变(SPG4型),10%患者携带ATL1基因突变(SPG3A型)。
34种为常染色体隐性遗传的痉挛性截瘫(AR-HSP),50%患者携带KIAA1840基因突变 (SPG11型),SPG15型是仅次于SPG11型的第2大最常见的AR-HSP。
5种为X-连锁遗传的痉挛性截瘫(XL-HSP),已确定有3个致病基因L1CAM(SPG1型),PLP1(SPG2型)及SLC16A2(SPG22型)与该类型的痉挛性截瘫有关。
1种母系遗传特征的痉挛性截瘫[7]。
遗传学特点
AD-HSP
在19种类型的AD-HSP中,11个相关的致病基因已经被确定,见表1。在AD-HSP家系中,70%~80%表现为单纯型HSP,并且40%~45%是由编码Spastin蛋白的SPAST基因突变引起的,SPG4型在散发病例中约占10%左右。ATL1基因突变是导致AD-HSP的第2大常见原因,并且是早发型AD-HSP最常见的原因。REEP1基因突变引起的SPG31型约占AD-HSP的5%。
AR-HSP
34种AR-HSP临床表现形式不尽相同,且大多数表现为复杂型HSP。AR-HSP在近亲结婚的人群中频率增高,目前26个亚型的致病基因已确定。SPG11型相关的基因突变是导致AR-HSP最常见的原因,占所有AR-HSP的20%,如果出现TCC和精神损伤该比例提高到60%~80%。ZFYVE26基因突变是导致AR-HSP的第2大原因。HSP亚型中经常出现痉挛性截瘫伴TCC的表型,目前至少有9个位点(SPG1,SPG7,SPG11,SPG15,SPG18,SPG21,SPG32,SPG46和SPG47)都与合并TCC的HSP有关[17],见表2。
XL-HSP
5种HSP遵循X染色体连锁遗传特征,其中已经发现3种L1CAM(SPG1型),PLP1(SPG2型)和SLC16A2(SPG22型)基因突变与XL-HSP有关,见表3。
母系遗传特征的痉挛性截瘫
目前在一个携带ATP6基因m.9176T>C突变家系的5个成员中出现晚发性痉挛性截瘫样疾病,生化检测结果显示线粒体出现明显损伤。该突变之前已被证明与其他疾病如亚急性坏死性脑脊髓病有关。这些结果表明修饰因子在疾病的表型和表现上起着重要作用,同样证明线粒体DNA(mtDNA)多态性与HSP的发生、发展有关[7]。
表1 19种AD-HSP的基本情况[7]
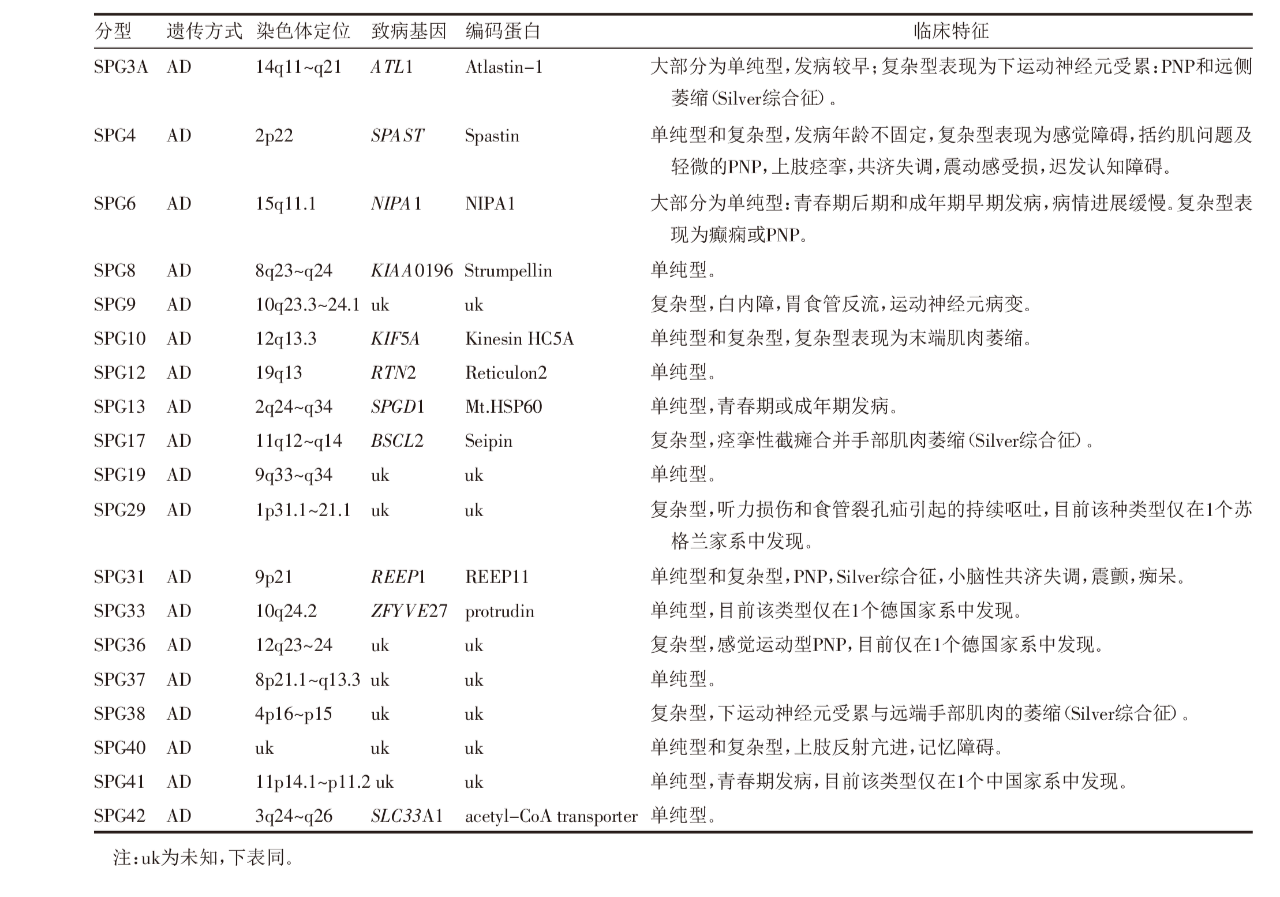
表2 34种AR-HSP的基本情况[7]
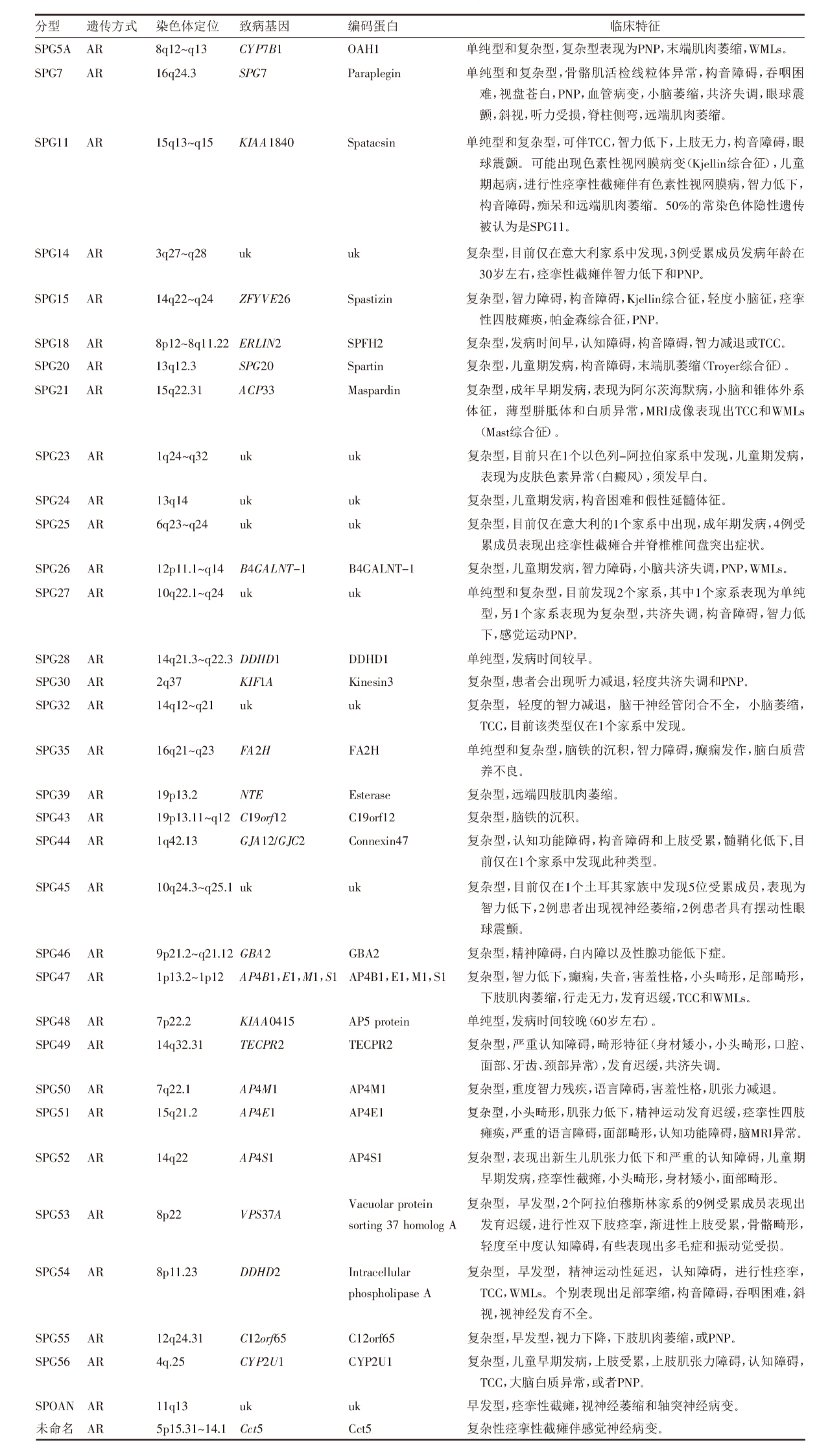
表3 5种XL-HSP的基本情况[7]
临床表现
遗传性痉挛性截瘫(HSP)患者可在婴幼儿时期时起病,亦可成年起病甚至老年起病,男女均可患病,男性多于女性,但也有报告女性多于男性。起病初期常表现为双下肢僵硬、无力行走易跌倒,逐渐发展成双下肢痉挛性截瘫,不能行走,需要拐杖、轮椅。不同遗传分型的HSP患者的临床表现、病情严重程度及病程进展各不相同,且同一家系内的不同患者其病情也不尽相同。许多研究表明:
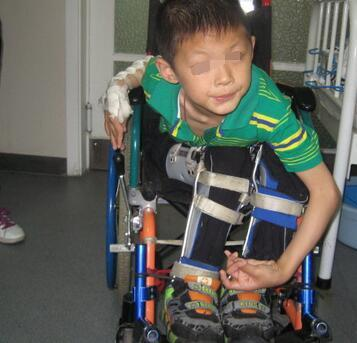
图1 HSP患者无力行走,需坐轮椅[9]
单纯型HSP患者的主要表现为:双下肢痉挛性截瘫、肌张力增高、腱反射亢进等、肌力正常或稍减退、双下肢感觉减退、括约肌功能障碍、足畸形、肢体远端肌萎缩、双上肢轻微的共济失调、踝反射消失等。另有有报道发现:约10%~65%的患者有感觉障碍,多表现为下肢远端位置觉和震动觉消失;约50%的患者有尿频、尿急现象;33%的患者出现足畸形,多发生在起病数年后;少数有下肢远端肌肉轻度萎缩,一般在起病10余年后出现;若有病例报道早期出现明显的肌肉萎缩,则应该警惕误诊的可能。少数患者上肢亦可受累,可在疾病早期出现,表现为上肢痉挛手运动障碍。但目前尚无脑神经受累的报道。复杂型HSP除了上述临床表现外还可伴有:癫痫间、智力障碍、锥体外系症状、共济失调、白内障、视神经萎缩、视网膜变性、鱼鳞病、周围神经病等。当前针对复杂性HSP的临床表现研究中发现:癫痫间类型与HSP之间的关系并不十分明确,有多种癫痫间类型发作的报道,包括肌阵挛发作、单纯部分性发作、复杂部分性发作和全面性发作等,且在同一家系内的不同患者可出现不同类型的癫痫间发作。智力障碍多表现为轻微的认知功能障碍,表现为近记忆力减退,注意力、计算力和理解力障碍等它可仅为HSP的伴随症状,亦可和其他伴随症状一起出现;多数患者从双下肢步行困难起病,而后出现眼部症状如眼球震颤、分离性斜视、假性眼外肌瘫痪;或者出现表情淡漠、动作减少、慌张步态等锥体外系的症状和体征,极个别的家系表现为痉挛性截瘫、肌张力不全、肌肉萎缩和智能减退同时存在,而且还有轻度感觉障碍、视神经萎缩和关节畸变等[8]。
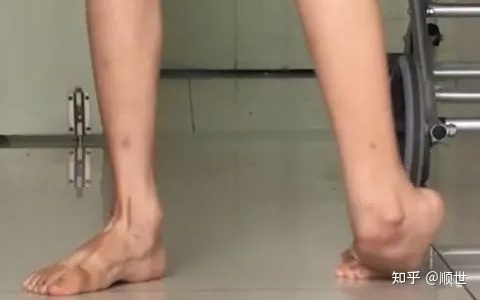
图2 HSP患者临床症状[10]
疾病诊断
HSP的初步诊断基于其缓慢进行性痉挛性双下肢无力的典型症状和(或)其他神经和非神经系统症状体征等,临床诊断通常参照HARDING的诊断标准:
①临床表现主要是双下肢无力、肌张力增高等上运动神经元受累症状,逐渐出现步态异常,进行性发展为双下肢痉挛性截瘫,部分病人可伴有尿频、尿急、认知障碍、癫痫发作、视力下降、锥体外系症状等;
②神经系统检查主要为锥体束征,下肢较明显;
③脑和脊髓CT或MRI检查多正常,但有部分病人可出现脊髓和(或)小脑萎缩,还可伴有胼胝体萎缩;
④多有家族史,符合常染色体显性遗传、常染色体隐性遗传、X-连锁隐性遗传或线粒体母系遗传,偶有散发病例;
⑤排除其他疾病所致的痉挛性截瘫,如脑瘫、多发性硬化症、肾上腺脑白质营养不良、运动神经元病等[5]。
由于HSP具有高度的临床及遗传异质性,临床表现复杂多变的同时缺乏特异性,且与其他神经疾病存在表型重叠,确诊仍有赖于基因检测,且需排除其他疾病。目前HSP基因检测阳性率只有45%~50%,可能原因有:HSP突变基因数量多且增长速度快,仍有许多未知基因尚未克隆;HSP基因突变可能表现为其他神经遗传性疾病;一些其他基因的突变与HSP复杂型表现有关;一些遗传性脊髓病的临床表现与HSP重叠。由于HSP的临床表型与基因型具有一定关联性,一些基因型具有相应的临床特征和影像学特点,临床医生可通过这些特点选择相应的基因进行检测。有学者根据发病年龄、家族史和遗传模式、临床特征及影像学异常,以靶向测序为一线检测手段,针对常见HSP基因型提出一种较为简易实用的诊断策略(见图3)。如若未检测到致病基因,可能存在拷贝数变异(copy number variants,CNV)如SPG4的SPAST外显子缺失,则需使用多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)进行复测,但该诊断策略尚未经过前瞻性测试,其临床价值还有待评估。若临床表型指向不明时可直接选择全外显子或全基因组测序的方法进行检测[11]。
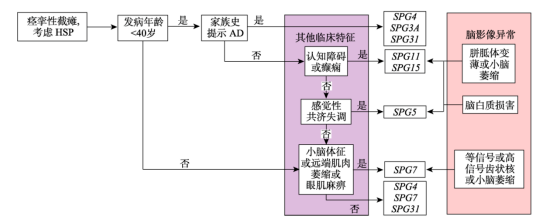
图3 常见HSP基因型的诊断策略[11]
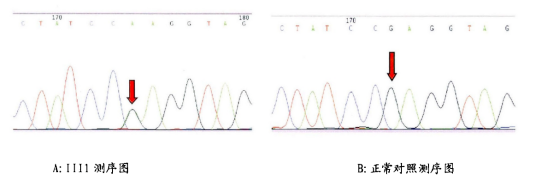
图4 HSP患者与正常基因测序图,该家系c.1685G>A[12]
动物模型
有学者发现,小鼠Borcs7基因的一个自发的截断突变,通过全基因组测序和遗传互补来确定,该突变除了影响溶酶体酶活性和内质网相关的内吞体小管融合的突变外,影响神经元细胞质内溶酶体迁移的突变也与神经退行性疾病相关,该突变导致BORC亚基BORCS7(C10orf32/干扰蛋白)的截断版本的表达,这导致了与人类HSP有许多相似的表型[7]。
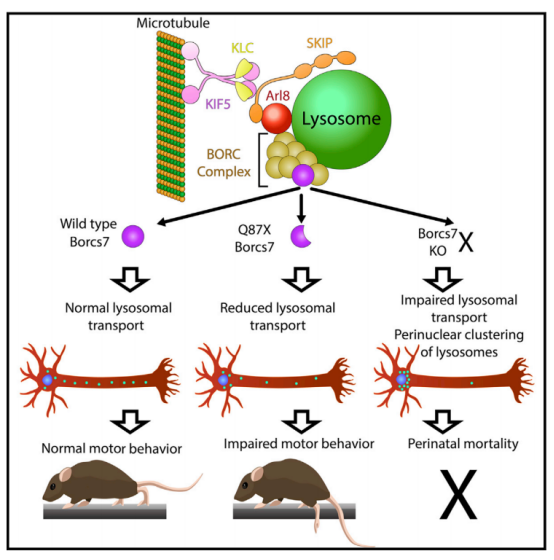
图5 溶酶体调节BORC复合物的Borcs7亚基的突变导致了小鼠的运动缺陷和营养不良的轴突病[13]
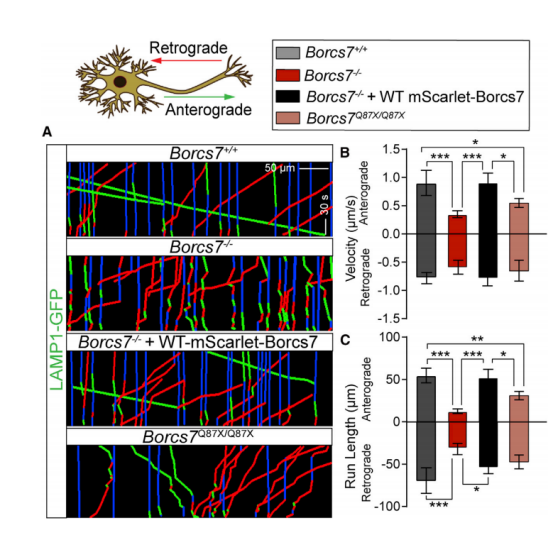
图6 Borcs7基因型对神经元溶酶体动力学的影响指示基因型轴突LAMP1-GFP动态。GFP标记的溶酶体在WT mScarletBORCS7拯救后显示正常的运动。(B和C)lamp1-gpf标记的囊泡的速度(B)和运行长度(C)。数据代表平均± SEM。*p < 0.05;**p < 0.01;***p < 0.001;与Tukey的后测试的单向方差分析[13]。
治疗方式
目前尚无特异疗法可以预防、延缓、逆转HSP,临床上多采用药物,理疗及手术等方法来缓解HSP患者的症状,提高其生活质量。
药物治疗
目前临床多采用左旋多巴、苯海索(安坦)、丁螺环酮、地西泮、氯苯丁氨酸、胞二磷胆碱、辅酶A、ATP等药物进行治疗。AdelineVanderver报道用左旋多巴/卡比多巴和沙丙蝶呤治疗SPG11型HSP有肯定疗效。
有学者曾报道1例中药结合针灸治疗HSP:取穴百合、双侧合谷、人中等,再辅以按摩推拿,一疗程15天,明显改善了肌痉挛、肌萎缩等症状。有学者报道了1例用单纯电刺激治疗HSP:给予患者双侧股四头肌电刺激,2~3次/周,连续治疗3个月,痉挛步态明显得到了改善[15]。
物理治疗对HSP患者ROM(关节活动度)和肌力的保持及改善都十分必要,并且能增强心血管系统耐受性,但不能阻止HSP病情进展。针对HSP患者的具体临床症状可以有选择地开展伸展训练、肌力训练和有氧训练,机器人步态训练和踝-足矫形器可用于加强功能性步态等,这些训练治疗可减轻肌腱炎、滑囊炎等并发症;提高肌肉的力量,减缓肌肉萎缩进程;改善心血管系统的适应性,减轻疲劳并提高耐力。

HSP患者接受物理治疗[14]
手术治疗
近几年国内外针对肢体严重畸形的HSP患者开展了矫形手术治疗,目的在于纠正长期痉挛造成的固定畸形,延长生存期,提高生活质量。有国外学者报道大部分患者可行跟腱和绳肌延长,以及内收肌松解手术。马凯等报道对4例单纯型HSP患者选择性切断L2-S1脊神经后根,术后患者的下肢痉挛症状明显缓解,步行距离延长,生活质量明显改善。近期国内外开展了周围神经缩窄术(selective peripheral neurotomy,SPN),逐渐用于治疗肢体远端肌肉痉挛状态明显的HSP患者。SPN的治疗机制是通过缩窄支配痉挛肌肉的运动神经末梢来减少神经纤维数量,达到降低神经冲动传导和减低牵张反射环路的兴奋性的目的,从而缓解肌肉痉挛程度,并改善肢体的运动功能[15]。
基因治疗
有学者报道将人胎儿脐带间充质干细胞通过鞘内注射和静脉输注方式移植到2例HSP患者体内,移植治疗后随访1年临床症状均有明显好转。基因治疗的主要步骤包括靶细胞和治疗基因的选择,可将外源性目的基因通过脂质体法、微粒轰击法和显微注射法等方法直接导入患者体内,使其进入目的细胞并在其内表达。还可以用逆转录酶病毒作载体将外源性正常基因导入靶细胞内,以弥补靶细胞的基因缺陷。再将其回输,使上述外源性基因在患者体内表达,达到基因治疗目的。
声明:本文收集归纳的信息,如有错漏,欢迎批评指正!
参考文献及照片来源:
[1]http://www.360doc.com/content/22/0124/22/36523718_1014763222.shtml
[2]詹飞霞,曹立.遗传性痉挛性截瘫4型发病机制的研究进展[J].上海交通大学学报(医学版),2017,37(09):1282-1285+1281.
[3]戎天艺,陈生弟.遗传性痉挛性截瘫的临床表现与基因诊断策略[J].诊断学理论与实践,2011,10(02):175-178.DOI:10.16150/j.1671-2870.2011.02.020.
[4]杨迪,高正玉,王强,张祎辰,安贝贝,潘晓娜.遗传性痉挛性截瘫的综合性认识与治疗现状[J].青岛大学学报(医学版),2020,56(04):500-504.
[5]耿挺. 科学家发现遗传性痉挛性截瘫致病新机制[N]. 上海科技报,2021-12-29(006).DOI:10.28704/n.cnki.nshkj.2021.001529.
[6]https://mp.weixin.qq.com/s/sD-Qcj-194MDOBzeYNJRxA
[7]禹文茜,段文元,鞠吉峰,王同建,朱萌,黄晶.遗传性痉挛性截瘫临床诊治与基因分型[J].国际生殖健康/计划生育杂志,2014,33(03):191-196.
[8]陈昕,赵国华,唐北沙.遗传性痉挛性截瘫的病理、遗传学、发病机制和临床的研究进展[J].临床神经病学杂志,2006(01):70-72.
[9]https://zhuanlan.zhihu.com/p/365296786
[10]https://zhuanlan.zhihu.com/p/400715124
[11]何秉涛,黄美欢,贠国俊,黄子薇,曹建国.遗传性痉挛性截瘫基因分型、分子诊断和治疗的研究进展[J].中国优生与遗传杂志,2022,30(03):516-519.DOI:10.13404/j.cnki.cjbhh.20220224.020.
[12]巫嫚. 遗传性痉挛性截瘫的临床分析[D].广西医科大学,2015.
[13]John N. Snouwaert,Rachel J. Church,Leigh Jania,MyTrang Nguyen,Matthew L. Wheeler,Andrew Saintsing,Piotr Mieczkowski,Fernando Pardo Manuel de Villena,Diane Armao,Sheryl S. Moy,Damaris N. Lorenzo,Beverly H. Koller. A Mutation in the Borcs7 Subunit of the Lysosome Regulatory BORC Complex Results in Motor Deficits and Dystrophic Axonopathy in Mice[J]. Cell Reports,2018,24(5).
[14]https://zhuanlan.zhihu.com/p/400715124
[15]张雷,徐爱军.遗传性痉挛性截瘫研究进展[J].临床荟萃,2013,28(03):347-349.