

AAV8基因疗法治疗肝脏代谢疾病里程碑式进展!CN患者摆脱12H光疗重获自由!
近日,Généthon宣布其在研基因疗法GNT0003治疗Crigler-Najjar综合征的1/2期临床试验结果在《新英格兰医学杂志》上发表。试验结果显示,这一基因疗法具有良好的安全性,并且在最高剂量表现出疗效。值得一提的是,这是在肝脏代谢疾病中首次通过临床试验证明了基因疗法的疗效。目前这一临床试验的关键性部分已经展开。

图1 来源:《新英格兰医学杂志》
Crigler-Najjar综合征是一种罕见的遗传性代谢疾病,由UGT1A1缺乏引起,导致游离胆红素在血清和身体所有组织中潜在致死性蓄积,可在大脑中产生毒性,有导致神经损伤和死亡的风险。GNT0003药物将AAV8载体与UGT1A1基因拷贝结合在一起,治疗的目标是恢复肝脏的功能,能够重新产生UGT1A1酶来负责分解胆红素。

该临床试验由Genethon赞助,汇集了来自意大利、荷兰和法国的研究人员,研究Genethon的GNT 0003候选基因疗法的有效性和安全性。该研究调查了17名年龄在21至30岁之间患有严重Crigler-Najjar综合征的女性。结果显示,该药恢复了UGT1A1基因的表达,使胆红素水平平均从351μmol/l降至149μmol/l。这种药物通过单次静脉注射将胆红素水平降低到毒性阈值以下,以至于三名接受最高剂量治疗的患者在过去18个月或更长时间内能够停止使用光疗,胆红素水平仍然维持在毒性水平之下。然而,接受低剂量药物治疗的患者在16周内胆红素就超过了毒性水平。该研究还支持该治疗的安全性发现,未报告严重不良事件。头痛和肝酶水平改变是最常见的不良事件,后者用糖皮质激素治疗。
虽然此次研究规模很小,但在接受5×10^12 vg / kg剂量的患者中,GNT0003将UGT1A1活性恢复到允许暂停光疗的水平,并且疗效在治疗后18个月持续存在。未来在更大的、特征明确的患者队列中进行复制测试将是重要的。Genethon正在进行一项更广泛的研究,从今年1月开始,评估该疗法对10岁及以上患者的疗效和安全性。
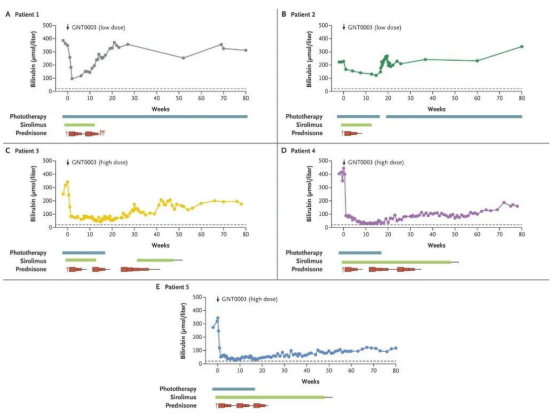
图3:GNT0003显著降低胆红素水平 来源:Généthon官网
传统疗法的巨大困境
GNT0003推动了一种治疗困难病症的潜在方法。迄今为止,没有药物被批准用于治疗CN综合征。光疗法仍然是严重CN患者长期护理的支柱,严重患者可能需要每天接受长达12小时的光疗,通过蓝色LED灯来分解身体无法分解的胆红素,但患者仍然存在胆红素脑病和神经系统后遗症的高风险,且光疗的有效性可能会随着年龄的增长而逐渐下降,导致更长的光疗持续时间。目前治疗这种疾病的唯一治愈方法是原位肝移植 (OLT) ,但面临着例如供体肝脏的可用性有限,需要终身免疫抑制以免发生排斥反应等局限性,因此极少有患者用此方法进行治疗。生活质量的限制和当前治疗的副作用清楚地表明了基因疗法等新方法的价值。
Genethon首席执行官Frederic Revah在一份声明中表示:“如果关键部分的结果证实了我们基因治疗Crigler-Najjar的有效性,我们将能够继续进行产品许可申请,并使患者可以使用这种治疗,为他们提供显着改善的生活质量。”
针对CN的另一基因疗法
目前另一个针对CN的临床试验是Astellas公司旗下Audentes研发的AT342,这是一种携带正常的UGT1A1基因的AAV载体。在该疾病的小鼠模型的临床前数据可知,Audentes使用单剂量的AAV-UGT1a1注入小鼠体内,小鼠体内的血清胆红素表现出持久的降低。血清胆红素水平是一个重要的临床检测标志,它可以直接检测AT342的易用性和可靠性。
图4 来源:药融云
基因疗法在肝脏代谢疾病的应用
由于肝在维持内环境稳态方面具有复杂而重要的特点,因此,许多疾病都源于肝脏。这些疾病包括遗传性疾病,如血色素沉着症,血友病a和 b,α1抗胰蛋白酶缺乏症,Wilson病,Crigler-Najjar综合征Ⅰ型,鸟氨酸转移酶缺乏症,IIa型家族性高胆固醇血症,以及纤维蛋白血症。因此,基因疗法治疗肝脏疾病也正在日益引起研究人员的关注。
iECURE研发GTP-506以治疗OTC缺乏症
鸟氨酸氨甲酰基转移酶(OTC)缺乏症是一种遗传性代谢病,由负责氨解毒的肝酶基因缺陷导致尿素循环障碍,临床主要表现为高氨血症,严重的可导致不可逆转的神经损伤、昏迷和死亡。目前尚无药物疗法治愈这种疾病,也难以消除致命性症状的风险,唯一的治疗方法是进行肝移植。GTP-506是iECURE的先导项目,通过AAV衣壳携带两种不同的有效载荷。GTP-506包含的两种载体之一是iECURE从Precision BioSciences获得的ARCUS核酸酶载体(GTP-506A),能够靶向良好表征的PCSK9基因位点进行基因编辑,另一种是治疗性供体载体(GTP-506D),能够插入健康的OTC基因以提供所需的疾病校正。该基因编辑疗法通过在PCSK9位点进行切割,然后在切割的位置插入治疗基因,为健康基因的永久表达提供了一种潜在途径。
此前,FDA已授予GTP-506孤儿药资格和罕见儿科疾病认定,用于治疗OTC缺乏症。2022年底,该公司宣布完成了6500万美元的A-1轮融资,使其筹集的资金总额达到了1.15亿美元。该资金将用于完成IND前活动、临床试验启动、接收iECURE用于OTCD先导研究产品的1/2期临床试验数据以及开发瓜氨酸血症1型(CTLN1)的IND前活动。
Vivet和辉瑞合作研发VTX-801以治疗WD
Wilson病是铜转运蛋白ATP7B基因突变引发的一种遗传疾病,发病概率约为1/40000人。Wilson患者的胆脏中代谢、排除铜元素的途径被破坏,导致组织中积累铜元素,引发组织毒性导致严重的肝脏损伤、神经系统症状,并可能导致死。2021年8月12日,Vivet Therapeutics 公司和辉瑞公司共同宣布,FDA已授予VTX-801快速通道资格,用于治疗威尔逊氏病(WD)。目前,VTX-801基因疗法已在临床前模型中得到了验证,并已获得FDA和欧盟委员会(EC)的孤儿药资格(ODD)认定。
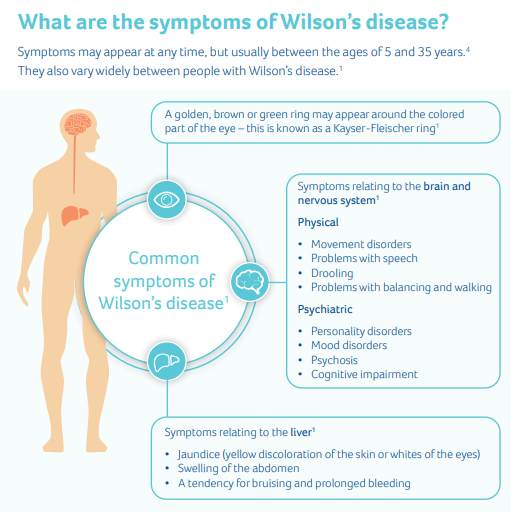
图5 来源:Vivet 官网
结语
基因治疗向来以攻克罕见病而见长,此次GNT0003治疗Crigler-Najjar的积极临床数据也为基因编辑疗法治疗肝脏代谢疾病的研究登上了一个更高的台阶。此外,也有更多的企业在加紧布局这一赛道,并获得了高额的融资用于临床阶段的推进,也有研究机构聚焦于尚未有药物被批准的适应症进行扩展,以全方位地克制多类疾病的共同源头—肝脏的恶化。相信随着基因疗法的进一步发展,将会有更多罕见病获得治疗乃至于治愈的机会。