

文献分享 | 杜氏肌营养不良症的基因补偿潜在疗法:转录适应
一、 研究概述
2025年2月12日,德国马普心肺研究所Didier Y. R. Stainier教授在Nature期刊发表标题为“Transcriptional adaptation upregulates utrophin in Duchenne muscular dystrophy”的研究论文。该研究主要揭示了在杜氏肌营养不良症(Duchenne muscular dystrophy, DMD)中,转录适应(transcriptional adaptation, TA)通过降解突变的DMD mRNA来上调utrophin(UTRN)的表达,并探索了通过反义寡核苷酸(ASOs)和核糖酶诱导遗传补偿的潜在治疗应用。

二、导读
杜氏肌营养不良症(Duchenne muscular dystrophy, DMD)是一种由DMD基因突变引起的X连锁隐性神经肌肉疾病。DMD基因编码的抗肌萎缩蛋白(dystrophin)是一种连接细胞骨架与细胞外基质的机械性连接蛋白,能够保护肌肉细胞免受收缩性损伤。目前,美国食品药品监督管理局(FDA)批准的DMD治疗方案包括针对特定缺失的外显子跳跃疗法、终止密码子通读化合物(如阿塔伦和Elevidys)以及递送微型抗肌萎缩蛋白的重组基因疗法。
Utrn基因编码的utrophin是DMD的同源基因,已被证明在携带移码或无义DMD突变的患者中,以及在mdx小鼠模型的骨骼肌肌膜中表达上调。这些突变导致提前终止密码子(PTC)的出现。然而,携带DMD基因框内缺失的患者并未表现出utrophin的上调。临床前研究表明,utrophin的表达与DMD疾病严重程度呈负相关。此外,与单基因突变的mdx小鼠相比,Utrn/Dmd双突变小鼠表现出更严重的表型。因此,utrophin的上调被认为是部分补偿抗肌萎缩蛋白缺失的机制,使其成为治疗DMD患者的有前景的策略。然而,utrophin上调背后的机制尚未明确,被认为与抗肌萎缩蛋白的丢失有关。
本研究开发了多种遗传工具,以研究携带移码或无义等位基因的DMD患者中utrophin上调的机制,并发现DMD突变信使RNA(mRNA)的降解通过一种新发现的细胞反应——转录适应(transcriptional adaptation, TA)在utrophin上调中发挥关键作用。此外,本研究还揭示了反义寡核苷酸(ASOs)和核糖酶在通过TA触发遗传补偿的新应用。在TA过程中,突变mRNA的降解会导致所谓的适应基因的转录增加。这一过程独立于蛋白质功能的丧失,并且在某些情况下可以实现功能补偿,可能解释了为什么关键基因中的移码或无义突变导致的PTC并不引起明显的表型。TA此前已在斑马鱼、秀丽隐杆线虫和小鼠细胞中被发现,但尚未在人类细胞中报道。
三、研究成果
1. DMD基因调控与UTRN上调的机制研究
本研究通过多种实验手段,系统地研究了DMD基因的转录调控机制及其对UTRN表达的影响。首先,作者研究了DMD第37号外显子(E37)的剪接调控,发现E37的3'剪接位点较弱且外显子剪接增强子密度较低,导致外显子频繁被跳过。通过使用3 µM的喜树碱(CPT)处理野生型人胚胎肾293T(HEK293T)细胞6小时,作者观察到E37外显子被跳过(图1a),表明其保留遵循共转录剪接的动力学模型。随后,使用1 µM的组蛋白去乙酰化酶抑制剂曲古霉素A(TSA)处理细胞24小时,发现TSA显著促进了E37外显子的保留(图1a)。这表明转录延伸速率对E37的剪接保留有重要影响。
为了进一步研究PTC对DMD转录本的影响,作者通过高保真Cas9技术在HEK293T细胞的DMD E37中引入一个20核苷酸缺失,导致PTC出现在其3'端上游118个核苷酸处(图1b)。实验结果显示,PTC的引入显著增加了E37的跳跃频率,表明PTC导致的转录本更易被无义介导的mRNA降解(NMD)。这一发现为研究DMD基因的转录调控机制提供了新的视角,并为基于转录调控的治疗策略奠定了基础。
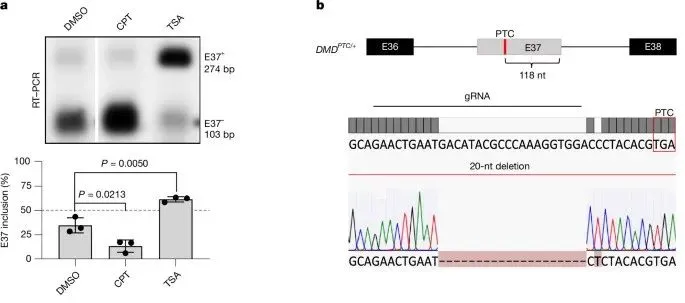
图1. 促进一个包含提前终止密码子(PTC)的可变剪接外显子的包含会触发DMD突变mRNA的降解以及UTRN的上调。
在DMD PTC/+细胞中,诱导包含PTC的E37外显子后,DMD mRNA水平显著降低(图1d),表明PTC导致的转录本被NMD降解。与此同时,UTRN mRNA水平显著上调(图1f),上调倍数约为1.5-2,与mdx小鼠模型相似。进一步检测发现,UTRN的前体mRNA(pre-mRNA)水平也增加,说明UTRN上调源于转录增加。尽管抗肌萎缩蛋白(dystrophin)水平未明显下降,但utrophin蛋白水平显著上调(图1g,h),这可能与抗肌萎缩蛋白的长半衰期有关。这些结果表明,UTRN上调是由于DMD突变mRNA降解引发的RNA反馈机制,而非抗肌萎缩蛋白的丢失。
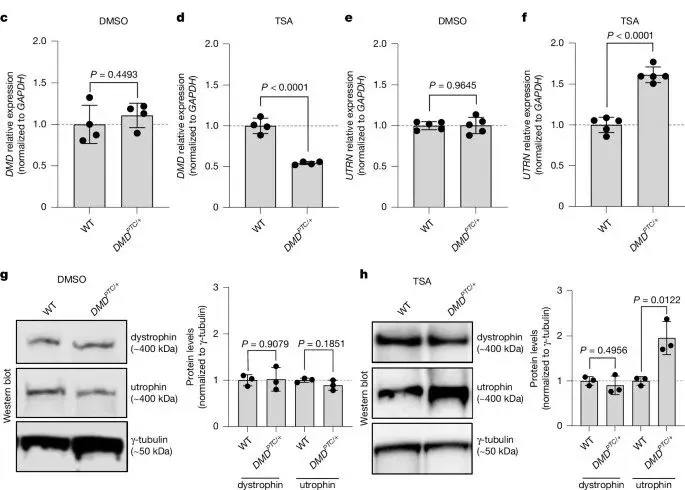
图1. 促进一个包含提前终止密码子(PTC)的可变剪接外显子的包含会触发DMD突变mRNA的降解以及UTRN的上调。
作者还发现,TSA处理对DMD E37外显子包含的影响在野生型(WT)和DMD PTC/+细胞中均呈现剂量和时间依赖性。随着TSA浓度的增加,DMD mRNA水平显著降低,而UTRN mRNA水平显著增加。在时间依赖性方面,TSA处理8小时后,DMD PTC/+细胞的DMD mRNA水平已显著低于WT细胞,而此时UTRN mRNA水平仅略有变化;然而,在TSA处理16小时后,UTRN mRNA水平显著上调(约1.5倍)。这些结果表明,UTRN mRNA水平的变化是由DMD突变mRNA的降解引起的。此外,作者通过洗脱TSA并让细胞恢复48小时,逆转了TSA的影响。恢复后,DMD E37的包含水平降低,DMD和UTRN mRNA水平也恢复正常。
这些发现揭示了DMD基因的转录调控机制及其对UTRN表达的影响,为理解Duchenne型肌营养不良症(DMD)的病理机制和开发潜在治疗方法提供了重要见解。
2. NMD阻断与DMD过表达对UTRN上调的影响
为了研究无义介导的mRNA降解(NMD)在DMD PTC/+细胞中的作用,作者通过敲低关键NMD蛋白UPF1和SMG6,发现阻断NMD后,DMD PTC/+细胞中DMD突变mRNA水平上升(图2a),而UTRN在mRNA(图2b)和前体mRNA(图2c)水平上的上调现象消失。相比之下,野生型(WT)细胞中阻断NMD对UTRN水平无影响,但导致DMD mRNA显著下降(图2a)。这些结果表明,DMD突变mRNA的降解是触发UTRN上调的关键,提示转录适应(TA)可能参与其中。
进一步地,作者在经TSA处理的DMD PTC/+细胞中过表达了DMD基因,以验证UTRN上调是否由TA引起。结果显示,抗肌萎缩蛋白的过表达并未影响UTRN mRNA或前体mRNA的上调(图2d–g),表明UTRN上调是由TA而非蛋白质反馈效应引起的。这进一步证实了DMD突变mRNA的降解在UTRN上调中的关键作用。
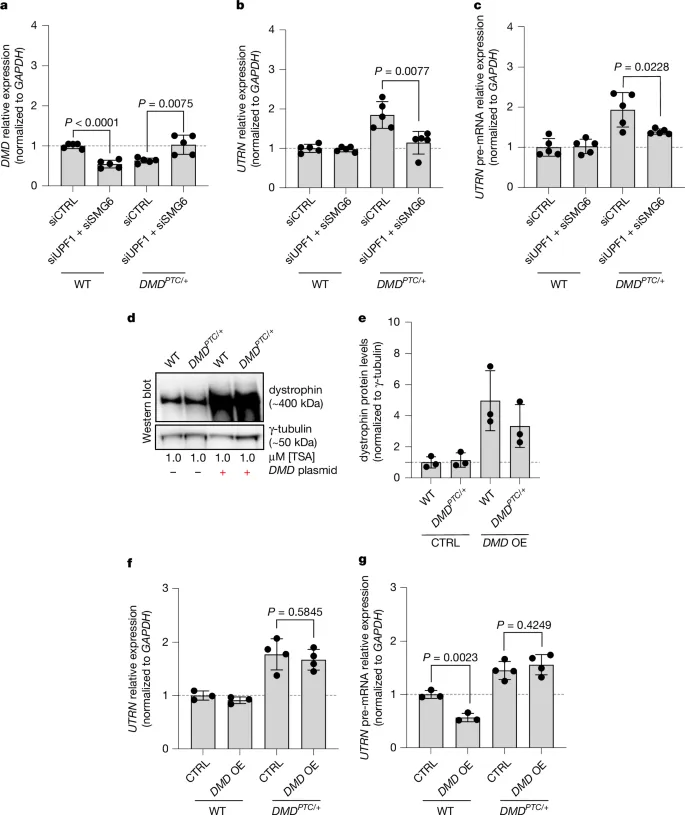
图2. 阻断NMD(无义介导的mRNA降解)可以逆转UTRN的上调,而过表达DMD则不能。
此外,作者分析了DMD患者的iPS细胞分化而成的肌管细胞的RNA测序数据,重点关注存在E51缺失(DMD2)的样本。这些细胞表现出UTRN上调,且在突变纠正后UTRN上调减少,提示TA的存在。作者还发现其他基因如ANO5、ACTA1、SERAC1、LIN28B和PPP1R14C等上调,这些基因直接或间接参与肌肉功能,表明它们可能通过上调来补偿抗肌萎缩蛋白的缺失。在mdx突变骨骼肌中,Acta1也显示出上调趋势,尽管不显著。这些发现表明,除了UTRN,其他基因也可能通过TA机制对DMD突变mRNA降解做出响应,从而在DMD病理过程中发挥补偿作用。
3. DMD突变mRNA降解对UTRN表达的调控作用
作者通过构建和转染DMD迷你基因,研究了DMD突变mRNA降解对UTRN表达的影响。首先,作者构建了包含不同外显子和内含子组合的DMD迷你基因,并引入无义突变生成DMD PTC迷你基因。这些迷你基因被转染到多种细胞系中,包括WT HEK293T、HAP1、HeLa细胞和肌管细胞。结果表明,转染DMDPTC迷你基因后,外源DMD mRNA水平显著降低,表明PTC导致的转录本降解(图3a)。
同时,UTRN mRNA水平在这些细胞中显著上调,而转染DMD WT迷你基因或空载体的细胞未观察到此现象(图3b-d)。这一现象在大多数细胞系中均有体现,但HeLa细胞除外。此外,某些特定PTC迷你基因(如DMD E338X)仅在HAP1和HeLa细胞中引起UTRN mRNA水平上调(图3b)。这些结果表明,DMD突变mRNA的降解能够触发UTRN上调,且这种反应因细胞类型和PTC位置的不同而存在差异。
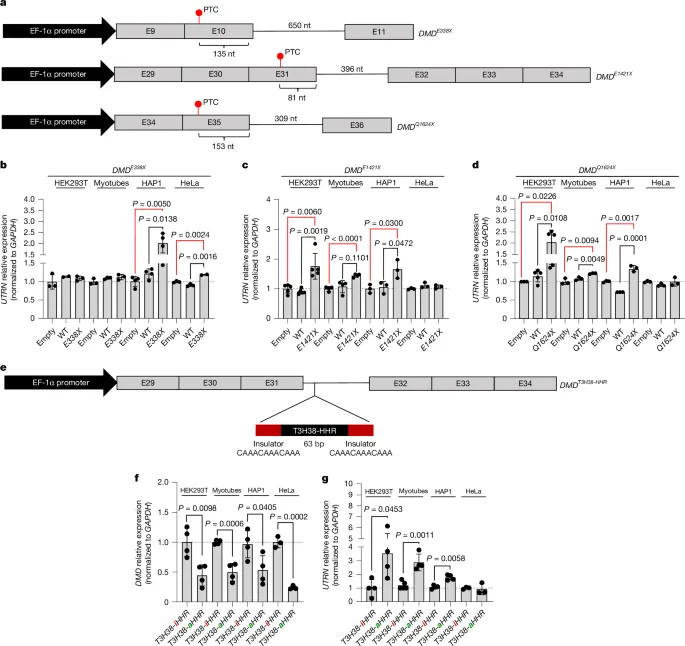
图3. 由DMDPTC迷你基因以及含有自切割核糖酶的DMDWT迷你基因诱导的UTRN上调。
进一步地,作者在DMD WT迷你基因的内含子中插入改良的锤头状核糖酶(T3H38-HHR),开发了一种不降低抗肌萎缩蛋白水平而降解DMD RNA的方法(图3e)。实验中,转染DMDT3H38-aHHR构建体的细胞显示出外源DMD mRNA水平的显著降低,而转染DMD T3H38-iHHR构建体的细胞则未受影响(图3f)。同时,转染DMD T3H38-aHHR构建体的细胞中UTRN mRNA水平显著增加(图3g),表明DMD mRNA降解是UTRN上调的主要触发因素。
然而,在HeLa细胞中,转染DMDT3H38-aHHR构建体并未导致UTRN mRNA水平上调(图3g),这与DMDE1421X迷你基因的结果一致(图3c)。这些发现表明,DMD迷你基因mRNA/pre-mRNA的降解即使在没有内源性抗肌萎缩蛋白丢失的情况下,也能导致UTRN mRNA水平的增加,进一步证实了DMD mRNA降解是DMD PTC/+细胞中UTRN上调的主要触发因素,并支持转录适应(TA)是某些DMD患者中UTRN上调的机制。
4. DMD突变与UTRN上调的关联及其治疗潜力
作者测量了来自4名DMD患者的肌管细胞中的DMD和UTRN mRNA水平,这些患者携带的突变均可能导致提前终止密码子(PTC)。结果显示,与健康对照组相比,所有DMD患者来源的肌管细胞中DMD mRNA水平降低,而UTRN mRNA水平显著增加(图4a,b)。此外,DMD患者肌管细胞中UTRN前体mRNA水平也增加,表明UTRN上调是由于转录增加所致。这些结果进一步支持DMD mRNA降解在UTRN上调中的关键作用。
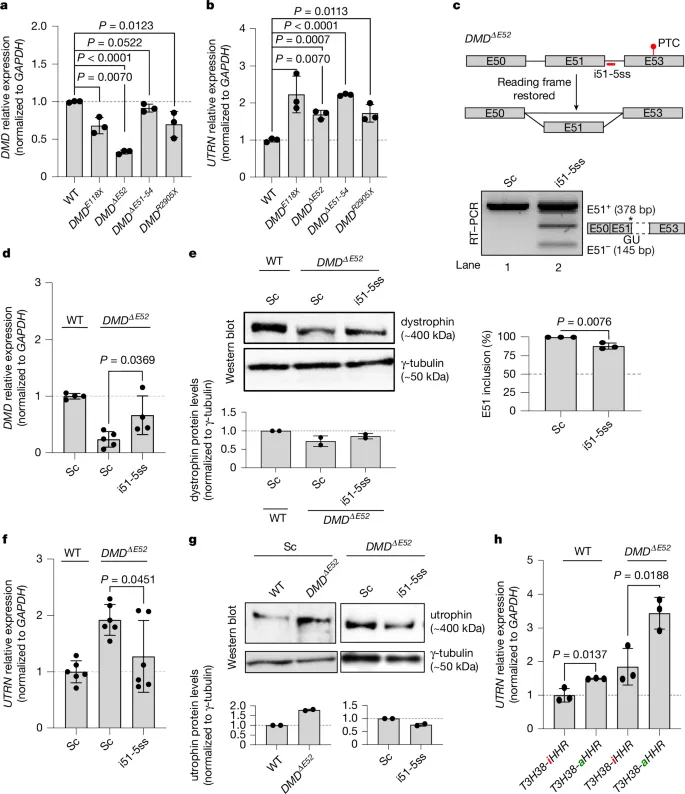
图4. 来自DMDPTC患者的肌管细胞表现出UTRN的上调,而在DMDΔE52肌管细胞中恢复DMD的阅读框则减少了这种上调。
作者尝试通过外显子跳跃恢复DMDΔE52肌管细胞中的DMD阅读框。他们设计了一种类似eteplirsen的ASO(i51-5ss),用于促进外显子51的跳跃(图4c)。结果显示,这种ASO显著增加了DMD突变肌管细胞中的DMD mRNA水平(图4d,e),但同时降低了utrophin的上调(图4f,g),可能抵消了恢复阅读框的部分有益效果。
随后,作者利用含有自切割核糖酶(DMDT3H38-HHR)的DMDWT迷你基因,尝试在DMDΔE52肌管细胞中增强UTRN的上调。结果表明,转染活性核糖酶的DMDΔE52肌管细胞中UTRN mRNA水平显著增加,而转染非活性核糖酶的细胞则无此现象(图4h)。这表明通过核糖酶诱导的DMD mRNA降解可以进一步上调UTRN,为DMD治疗提供了新的可能性。
5. Splice-switching ASOs可触发UTRN上调
作者采用剪接转换ASOs(反义寡核苷酸)靶向DMD基因的两个框外外显子E6(i6-5ss)和E52(i52-5ss)的5'剪接位点,诱导其跳跃(图5a)。这种跳跃通过破坏阅读框引入提前终止密码子(PTC),从而触发UTRN上调。实验中,与对照ASO相比,E6和E52的跳跃效率接近100%(图5b),并导致DMD突变mRNA降解及UTRN上调(图5c,d)。
作者进一步推测,部分外显子跳跃(25-50%)也能在不显著降低DMD mRNA或蛋白水平的情况下触发UTRN上调。结果表明,部分跳跃确实导致UTRN上调(图5g,h),且DMD mRNA(图5f)和蛋白水平(图5h)未显著降低,这可能是因为全长异构体比跳跃异构体更为常见(图5e)。
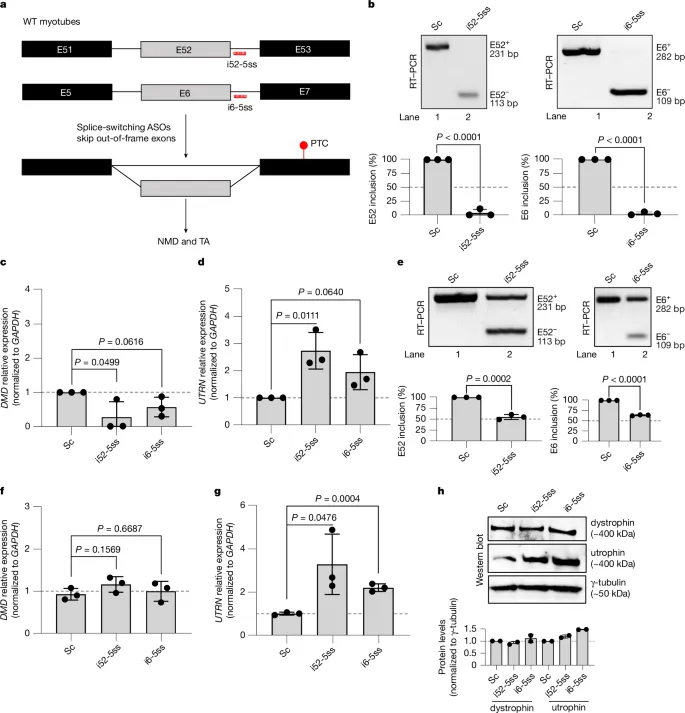
图5. 通过剪接转换反义寡核苷酸(ASOs)在DMD中引入提前终止密码子(PTC)会触发UTRN的上调。
此外,作者使用之前用于恢复DMD ΔE52肌管细胞阅读框的i51-5ss ASO,发现其也能在WT肌管细胞中破坏阅读框并触发UTRN上调。这一结果进一步支持了转录适应(TA)在UTRN上调中的作用。
四、讨论
自mdx小鼠模型被用于研究Duchenne型肌营养不良症(DMD)以来,utrophin的上调被发现可以补偿抗肌萎缩蛋白(dystrophin)的缺失。此后,utrophin上调成为治疗该疾病的主要策略之一。然而,utrophin上调的机制尚不清楚,通常被认为是由抗肌萎缩蛋白丢失引起的。本研究通过四种不同的方法提供了证据,表明DMD mRNA/pre-mRNA的降解足以上调UTRN,并且NMD(无义介导的mRNA降解)对于DMDPTC/+细胞中UTRN的上调是必需的。
转录适应(TA)作为一种遗传补偿机制,最初是在研究斑马鱼突变表型与反义(morpholino)诱导表型差异时发现的。进一步的研究揭示了TA在秀丽隐杆线虫(C. elegans)中的存在,并确认突变mRNA的降解在斑马鱼、秀丽隐杆线虫以及小鼠细胞中触发TA的关键作用。然而,此前TA尚未在人类中被报道。本研究首次展示了人类中的TA现象及其在遗传性疾病中的潜在作用。研究发现,携带E52缺失的DMD患者肌管细胞表现出UTRN上调。使用类似eteplirsen的ASO跳过E51可减少UTRN上调。对于这些DMD患者,使用含有自切割核糖酶的DMD和/或UTRN迷你基因可能进一步上调UTRN,与eteplirsen治疗产生协同效应。当然,大多数DMD患者可能都会从这些含有自切割核糖酶的迷你基因中受益。
错义突变在许多人类遗传疾病中比移码或无义突变更常见,而移码或无义突变常导致mRNA降解。在某些疾病(如镰状细胞贫血、马凡综合征和由MYH7突变引起的肥厚型心肌病)中,错义突变的影响比无义突变更严重。通过TA实现的功能补偿可能解释了为什么无义突变的报告频率低于错义突变,因为它们可能导致更温和的表型。
本研究展示了剪接转换ASOs的新应用,通过触发突变mRNA降解实现功能补偿。此外,含有自切割核糖酶的迷你基因提供了另一种诱导TA的方法,可能适用于错义突变患者以及移码或无义突变患者,因为增加的RNA降解可以进一步上调补偿基因。利用DMD E37的可变剪接,本研究构建了一个可诱导的DMD突变mRNA降解模型,为诱导内源性mRNA降解提供了新工具。
近年来的研究表明,可变剪接在多细胞生物中极为普遍,约95%的人类基因发生可变剪接,且约20%的剪接事件对转录延伸敏感。因此,这种可诱导的剪接依赖系统有望在广泛基因中触发mRNA降解,并观察其随时间的变化。此外,诱导包含PTC的延伸敏感外显子(“毒外显子”)或通过剪接转换ASOs引入PTC,也是触发mRNA降解的有前景的方法,且无需修改基因组。另一种可能的TA触发策略是利用最近发现的“邻近诱导核酸降解剂”,通过小分子靶向降解RNA。未来还可探索RNA干扰或其他RNA降解方式是否能触发TA或类似TA的响应。总体而言,这些发现突出了TA作为人类遗传稳健性机制的重要性及其与遗传性疾病的相关性,有助于设计新的治疗策略,利用或不干扰TA触发的功能补偿。
来源:BioMedDaily
【声明】本文为转载文章,本平台仅作分享、传递信息,版权归原作者所有,如有侵权,请联系删除。